









The lipoprotein(a) (Lp(a)) molecule consists of an apolipoprotein (Apo) B-containing LDL-like segment and a plasminogen-like glycoprotein Apo(a) segment. An increased risk of cardiovascular disease, including acute coronary syndrome, ischaemic stroke and aortic stenosis, in patients with high plasma Lp(a) has been demonstrated in clinical studies, genome-wide association studies and Mendelian randomisation studies.1–4 The increased risk is often explained by the enhanced progression of atherosclerosis in the arterial system through the proatherogenic and anti-fibrinolytic nature of Lp(a) in combination with other factors, including age, sex, ethnicity, comorbidities and LDL cholesterol level. The independent association between elevated Lp(a) and an increased risk of cardiovascular events has been demonstrated, therefore the role of residual risk in patients with statin-based conventional therapy has been emphasised.2 The level of Lp(a) is established during childhood and is mostly consistent throughout the lifetime because it is regulated primarily by the LPA gene locus. However, several therapies, including proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, inclisiran and antisense oligonucleotides, have been reported to reduce the plasma level.5–8 Approximately 20–30% of people have elevated Lp(a), so more clinical attention is required.9
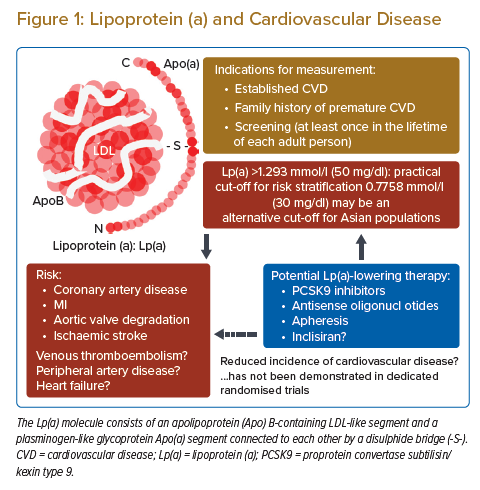
In this review, we summarise the clinical impact of elevated Lp(a), the evaluation of Lp(a) in daily practice, and the potential therapeutic approaches for high Lp(a) (Figure 1).
Epidemiology of High Plasma Lp(a)
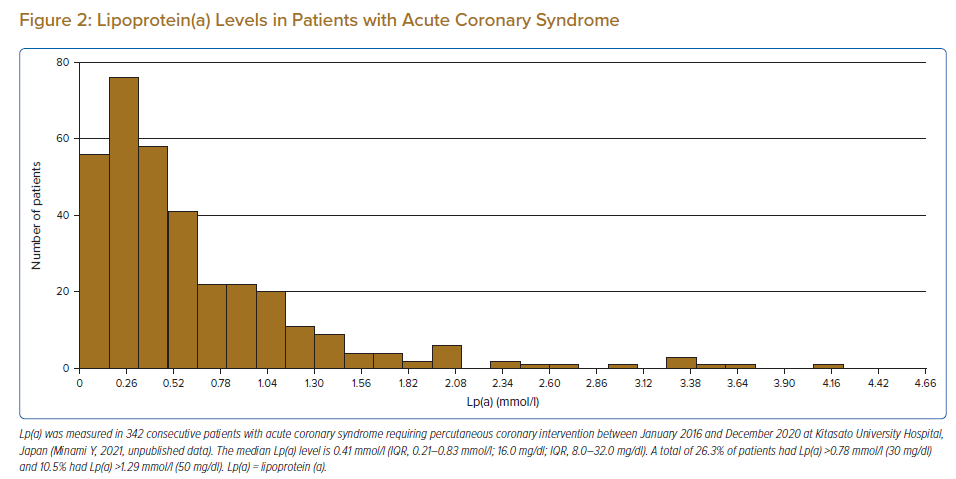
The distribution of plasma Lp(a) level is skewed and ranges widely between individuals, with no sex differences.3 Plasma concentration is determined primarily by the LPA gene locus, without significant acquired or environmental influences.10 Patients with established cardiovascular disease had higher levels of Lp(a) (Figure 2). There are ethnic differences in median Lp(a) level between countries caused by the difference in the prevalence of LPA single nucleotide polymorphisms and Apo(a) isoforms.11,12 The median Lp(a) level in the general Asian population is 0.21–0.36 mmol/l, which is lower than that in black (1.01 mmol/l), Hispanic (0.49 mmol/l) and Arab (0.39 mmol/l) populations.11,13–15
Cut-off of Lp(a) for Risk Stratification
Several practical cut-offs for Lp(a), including 0.78 mmol/l (30 mg/dl) and 1.29 mmol/l (50 mg/dl) for the risk stratification of coronary artery disease (CAD) and ischaemic stroke, have been proposed. The Copenhagen City Heart Study (n=7,524) found that Lp(a) >0.78 mmol/l is associated with an increased risk of MI in a dose-dependent manner, with an adjusted HR of 1.2 (95% CI [0.9–1.6]) for Lp(a) 0.13–0.75 mmol/l (5–29 mg/dl), 1.6 (95% CI [1.1–2.2]) for Lp(a) 0.78–1.97 mmol/l (30–76 mg/dl), 1.9 (95% CI [1.2–3.0]) for Lp(a) 1.99–3.03 mmol/l (77–117 mg/dl), and 2.6 (95% CI [1.6–4.1]) for Lp(a) >3.03 mmol/l (117 mg/dl) versus for Lp(a) <0.13 mmol/l (5 mg/dl).3
A meta-analysis of 126,634 participants showed that the risk of MI started to increase steeply at Lp(a) >0.62 mmol/l, and curvilinearly increased according to the increase in Lp(a) and became significant at around 1.24 mmol/l.2 A genome-wide association study also showed a steep increase in risk at >0.78 mmol/l and an added risk at >1.22 mmol/l.16 Current guidelines in the US, Canada and Europe define 1.29 mmol/l (50 mg/dl) as a risk-enhancing factor.17–19
Globally, the estimated number of people with elevated Lp(a) is >1 billion. Approximately 10% of the general Asian population may have Lp(a) >1.29 mmol/l, compared to 15–30% of the global population.20 A recent report investigated ethnic differences in the HR of high Lp(a) (>1.29 mmol/l) for the incidence of MI.15 The HR in Chinese, Southeast Asian and South Asian populations is reported as 1.62, 1.83, and 2.14, respectively, while it is 1.36 in European and 1.67 in Latin American populations. Although few reports have focused on the practical cut-off point of Lp(a) for Asian populations, Lp(a) ≥0.78 mmol/l (30 mg/dl) may be more suitable for the Chinese population.20,21 Several studies have suggested that elevated Lp(a) remains a risk factor for cardiovascular disease, even in patients with LDL cholesterol <1.81 mmol/l.22–24 Meanwhile, a recent study reported that the association of elevated Lp(a) and the risk of cardiovascular disease is attenuated in a primary prevention setting at LDL cholesterol <2.5 mmol/l.25
Impact of Elevated Lp(a) on the Incidence of Cardiovascular Disease
Elevated Lp(a) is associated with an increased risk of premature onset of CAD, ischaemic stroke, peripheral artery disease (PAD) and aortic valve degradation.2,26–28 The association of elevated Lp(a) and an increased incidence of recurrent cardiovascular disease in patients treated with statin and percutaneous coronary intervention (PCI) has been also reported.29–32
Elevated Lp(a) and Coronary Artery Disease
In the general population, a significant increase in the risk of coronary death and non-fatal MI has been observed in people with Lp(a) >1.29 mmol/l (50 mg/dl).2 In patients with established CAD, elevated Lp(a) increases the risk of recurrent clinical events, particularly in those with LDL cholesterol ≥3.37 mmol/l.33,34 The increased risk of recurrent cardiovascular events, according to the increase in Lp(a), has been shown even in patients treated with statins.29 The increased incidence of CAD in people with elevated Lp(a) is often explained by the higher prevalence of coronary plaque through the effect of the proatherogenic properties of Lp(a) particles.35,36 The association of elevated Lp(a) with a greater number and degree of coronary stenosis on angiography, a larger number of stenoses on CT, and a greater change in the plaque-plus-media area on serial intravascular ultrasound has also been reported.35–37 Chieng et al. investigated the impact of Lp(a) and LDL cholesterol levels on the angiographic disease severity assessed using the SYNergy between percutaneous coronary intervention with TAXus and cardiac surgery (SYNTAX) score in patients with premature CAD admitted to hospital in Australia.38 They showed that patients with both elevated Lp(a) and elevated LDL cholesterol comprised the majority of patients in the highest SYNTAX tertile, while patients with non-elevated Lp(a) and non-elevated LDL cholesterol were predominant in the lowest SYNTAX tertile.38
Muramatsu et al. investigated the impact of Lp(a) and LDL cholesterol level on the prevalence of vulnerable coronary plaque using optical coherence tomography in a Japanese population.39 They noted an increased prevalence of thin-cap fibroatheroma in the culprit coronary plaque with an increase in Lp(a) level, particularly in patients with LDL cholesterol ≥2.59 mmol/l.39 The increased incidence of recurrent cardiovascular events after PCI in patients with elevated Lp(a) has been shown in a Japanese cohort with chronic kidney disease and diabetes.30,31
A recent report from Korea has also shown the impact of elevated Lp(a) on the increased incidence of recurrent ischaemic cardiovascular events including repeated revascularisation and ischaemic stroke after PCI.32 The authors showed that the HR of recurrent events in patients with Lp(a) ≥0.78 mmol/l (30 mg/dl) during a median follow-up of 7.4 years was 1.17 (95% CI [1.05–1.30]; p=0.004). They noted that the enhanced proliferation of smooth muscle cells in addition to accelerated atherosclerosis via elevated Lp(a) might be a contributing factor.
Elevated Lp(a) and Peripheral Artery Disease
A modest but significant association between Lp(a) and PAD was noted in a study of Mendelian randomisation.26 The authors showed that increases in genetically predicted Lp(a) were associated with an increased risk of PAD (OR 1.04 per 0.26 mmol/l increase in Lp(a)). They also found that the association was not attenuated in multivariable analyses, accounting for the association of these genetic variants with ApoB. Further studies are needed to confirm the impact of elevated Lp(a) on the increased incidence of PAD.
Elevated Lp(a) and Stroke
A recent meta-analysis showed a significant association between elevated Lp(a) and an increased risk of ischaemic stroke compared with control subjects.27 The association between elevated Lp(a) and the increased risk of ischaemic stroke was also demonstrated in Asian patients.40–42 The meta-analysis further showed that elevated Lp(a) was significantly associated with an increased risk of the large artery atherosclerosis subtype as well as an increased risk of intracerebral haemorrhage. Most of the studies included in the meta-analysis used 0.52–1.04 mmol/l (20–40 mg/dl) as the cut-off for elevated Lp(a).
A recent prospective and observational study investigated the relationship between acute ischaemic stroke and serum Lp(a) level in the Chinese population.41 The authors reported a significant difference in median serum Lp(a) between patients with acute ischaemic stroke and control cases (0.85 versus 0.39 mmol/l [33 versus 15 mg/dl]; p=0.000). The authors also reported that elevated Lp(a) was an independent factor for stroke, and serum Lp(a) ≥0.78 mmol/l (30 mg/dl) was associated with a 2.23-fold increase in acute ischaemic stroke when adjusting for other possible risk factors. The pathogenesis of an increased incidence of ischaemic stroke in people with elevated Lp(a) could be explained by advanced carotid atherosclerosis.43,44 The association between increased Lp(a) and a higher prevalence of vulnerable plaque characteristics in the carotid artery in patients with symptomatic carotid artery stenosis has been demonstrated in a previous study.45
Elevated Lp(a) and Aortic Valve Disease
The association between elevated Lp(a) and calcification and stenosis of the aortic valve has been reported in several studies.46,47 A study combining two prospective studies of the general population showed that elevated Lp(a) and the corresponding genotypes were associated with an increased risk of aortic valve stenosis in the general population, with Lp(a) >2.33 mmol/l (90 mg/dl) predicting a threefold higher risk.28 A recent study with serial observations using multiple modalities showed that patients in the highest Lp(a) tertile had a higher progression of valvular calcification, faster haemodynamic progression, and a higher risk of aortic valve replacement and death compared with patients in the lower tertiles.48 Increased alkaline phosphatase activity, hydroxyapatite, cell apoptosis and phosphorylation of signal transduction proteins have been proposed as a conceivable pathway of aortic valve degradation by elevated Lp(a).49 Although the ethnic difference in the effect of elevated Lp(a) on degradation of the aortic valve remains unclear, a recent study reported that there were no associations between Lp(a) level and the extent of aortic valve calcification in South Asian people, although the association was seen in white people and black people.50
Indications for Lp(a) Measurement
The potential of Lp(a) measurement as a tool for the stratification of risk of future cardiovascular events was shown in a study of 826 people in the general population.51 Elevated Lp(a) was associated with an increased risk of cardiovascular disease over a 15-year follow-up period: the adjusted HR for cardiovascular events was 1.37 per 1 SD higher level of Lp(a) (SD=0.83 mmol/l [32 mg/dl]) and 2.37 compared with the top fifth quintile. The evaluation of Lp(a) has been shown to improve the quality of risk stratification in the general population with borderline risk, defined using the Framingham risk score.25,52
Measurements of Lp(a) enable the reclassification of the risk of cardiovascular disease and facilitate shared decision-making for the initiation of treatment, especially in younger patients, particularly those with a family history of premature cardiovascular disease. Current guidelines from Europe and North America recommend measuring Lp(a) in patients with a family history of premature cardiovascular disease, with Lp(a) >1.29 mmol/l (50 mg/dl) considered a risk-enhancing factor.17–19 A Mendelian randomisation study showed that extremely elevated Lp(a) (>5.172 mmol/l [200 mg/dl]) is associated with a three- to fourfold increased risk of cardiovascular disease, representing a similar lifetime risk of cardiovascular disease in heterozygous familial hypercholesterolaemia.53
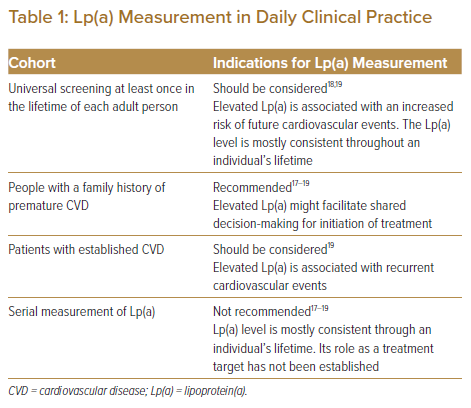
Although it remains unclear whether universal testing of Lp(a) is recommended for healthy people regardless of the presence of a family history, the potential causal association between Lp(a) and future cardiovascular events has been shown in several Mendelian randomisation studies, which could justify universal screening.1–4 Some of the current guidelines recommend Lp(a) measurement at least once in the lifetime of each adult person to identify people who may have inherited extremely high Lp(a) equivalent to the risk associated with familial hyperlipidaemia.18,19 In patients with established cardiovascular disease, elevated Lp(a) is associated with an increased incidence of recurrent cardiovascular events irrespective of LDL cholesterol.6,54 However, no randomised controlled trial has demonstrated a significant reduction in recurrent clinical events through lowering of Lp(a). Therefore, plasma Lp(a) cannot be a treatment goal in current clinical practice, and the serial measurement of Lp(a) is not recommended in guidelines.17–19 The possible indications for Lp(a) measurement in daily clinical practice are listed in Table 1.
Issues of Lp(a) Measurement
Lp(a) level may sometimes need to be interpreted with caution. Because of the wide range in the size of the Apo(a) segment and lipid content, the molecular mass of the Lp(a) particle also varies widely. The size of the Apo(a) segment depends on the number of kringle (KIV2) repeats, which is genetically determined.55 This may cause an ethnic difference in the size distribution of the Lp(a) molecule and raise difficulties in the application of results to patients of a different ethnicity.56 Although most currently available assays provide acceptable accuracy in the measurement of Lp(a) level to differentiate high-risk patients, the system of different assays still requires standardisation and appropriate calibration.1,56
Potential Approaches for Lowering Lp(a)
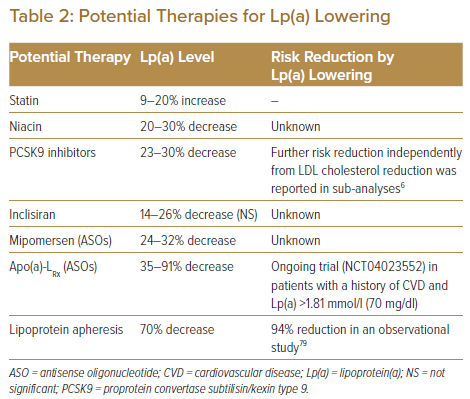
There have been no dedicated randomised clinical trials on the impact of Lp(a) lowering on the reduction of cardiovascular disease. Although the pharmacological mechanisms are unclear, a potentially significant decrease in plasma Lp(a) by several drugs has been reported.7,8,57 For primary prevention in people with intermediate risk, defined as a cardiovascular disease risk of 7.5–20% calculated using the Framingham risk score, elevated Lp(a) may be considered a risk-enhancing factor to start statin therapy.17,58 In high-risk or very-high-risk patients with LDL cholesterol ≥6.98 mmol/l on maximally tolerated statin treatment, it might be reasonable to consider more intensive treatment based on elevated Lp(a).59 In the setting of secondary prevention, the presence of a high Lp(a) level is strongly predictive of recurrent cardiovascular events and suggests the need for more intensive therapies, including PCSK9 inhibitors. Table 2 summarises the current status of potential therapies for Lp(a) lowering and subsequent reduction of cardiovascular events.
Statins
The role of statins in the primary and secondary prevention of cardiovascular events has been established. However, several prospective statin studies have shown a significant increase in plasma Lp(a) after starting statin therapy.34,60–64 A meta-analysis showed that statin increased Lp(a) by 8.5–19.5% from baseline, although it remains to be elucidated whether the statin-mediated increase in Lp(a) contributes to the increased risk of clinical events.65 Another meta-analysis found that statins may modify the association between Lp(a) and the risk of cardiovascular disease.29 In the analysis, Lp(a) level was shown to be more strongly associated with an increased risk of cardiovascular disease in patients with statins than in those with placebo.29
Niacin
Niacin, also known as vitamin B3, is an essential vitamin in the human body. Niacin can decrease total cholesterol, LDL cholesterol and triglyceride levels, and increase HDL cholesterol in large doses. Its lipid-lowering function is associated with the inhibition of hormone-sensitive lipase activity in adipose tissue, reduction of free fatty acid entry into the liver, and decrease in very-low-density lipoprotein secretion.66 Niacin also reduces Lp(a) levels by 20–30% by inhibiting the LPA promoter.23 However, HPS2-4THRIVE, a double-blind randomised trial, demonstrated that niacin failed to reduce cardiovascular events in patients with CAD. The addition of niacin to statin treatment resulted in significant increases in adverse effects, including disturbances in diabetes control, dysplasia, diarrhoea, myopathy, serious infection and skin ulcerations.67 The combination of niacin and statins in lipid-lowering therapy is no longer recommended in most countries.18
PCSK9 Inhibitors
PCSK9 inhibitors have been shown to reduce Lp(a) by 7–36%.54,57,68 This effect is consistent and reproducible for both alirocumab and evolocumab, although the pharmacological mechanism of reduction of Lp(a) by PCSK9 inhibitors remains unclear.69,70 In the FOURIER trial that included patients with cardiovascular disease and maximum tolerated dose of statin, evolocumab significantly reduced Lp(a) by a median of 26.9% at 48 weeks.54 Evolocumab has been shown to reduce the risk of cardiovascular events by 23% in patients with high Lp(a) and by 7% in those with low Lp(a). This indicates that the higher the baseline Lp(a), the greater the potential benefit of PCSK9 inhibitors in reducing cardiovascular events.54 A subanalysis of the Odyssey Outcomes trial showed that alirocumab-induced reductions of Lp(a) independently predicted lower risk of recurrent cardiovascular events, after adjustment for baseline concentrations of both LDL cholesterol and Lp(a) and demographic and clinical characteristics (i.e. a 0.03 mmol/l [1 mg/dl] reduction in Lp(a) with alirocumab was associated with a HR of 0.994 for recurrent cardiovascular events).6
Inclisiran
A potential for Lp(a) lowering by inclisiran has been reported although the pharmacological mechanism remains unclear. Inclisiran is a small interfering RNA (siRNA) molecule that reduces LDL cholesterol through the inhibition of PCSK9 synthesis. The safety and efficacy of inclisiran given twice a year by subcutaneous injection have been demonstrated.7,71,72 The current ongoing randomised trial ORION-4 (NCT03705234) will provide information on the effect of inclisiran on the incidence of cardiovascular events. In addition to the reduction of LDL cholesterol levels by 52.3% without serious adverse effects, inclisiran also reduced Lp(a) by 14–26%, although this did not reach statistical significance.7,73
Antisense Oligonucleotides
Mipomersen is an antisense oligonucleotide (ASO) that is designed to inhibit the synthesis of the ApoB100 protein and which produces a modest reduction in Lp(a). The percentage change in LDL cholesterol and Lp(a) with mipomersen therapy was −24.0% and −31.6%, respectively.74 Apo(a)-LRx is also an ASO, and it is rapidly and specifically taken up by hepatocytes and inhibits the synthesis of Apo(a).75,76 Apo(a)-LRx has been shown to reduce Lp(a) by 66–91% in a dose-dependent manner in healthy participants with elevated Lp(a), as well as in patients with established cardiovascular disease and elevated Lp(a).8,77
In a randomised trial of patients with established cardiovascular disease who had Lp(a) ≥1.55 mmol/l (60 mg/dl) on screening, Apo(a)-LRx at a dose of 20 mg weekly for 6 months resulted in an 80% reduction in Lp(a) level, and 98% of the patients on this dose attained an Lp(a) level <1.29 mmol/l (50 mg/dl).8 Another randomised trial compared the Lp(a)-lowering and anti-inflammatory effects of Apo(a)-LRx with PCSK9 inhibitors.78 In that study, the percent change in Lp(a) in patients with Apo(a)-LRx and PCSK9 inhibitors was −46.6% and −16.1%, respectively. Furthermore, potent and highly selective Lp(a) lowering by Apo(a)-LRx resulted in the reduction of inflammatory gene expression in circulating monocytes in patients with cardiovascular disease. In contrast, PCSK9 inhibitors did not alter Lp(a)-induced proinflammatory profiles.78 These findings support the potential benefit of improving clinical outcomes through a greater reduction of Lp(a) in patients with established cardiovascular disease. The current Phase III study (HORIZON; NCT04023552) will clarify the treatment effect of Apo(a)-LRx on clinical outcomes in patients with cardiovascular disease.
Lipoprotein Apheresis
Lipoprotein apheresis (LA) is the final escalating option to reduce blood LDL cholesterol levels in patients with severe hypercholesterolaemia, such as familial hypercholesterolaemia or other forms of hypercholesterolaemia resistant to or intolerant to lipid-lowering medication. LA should be considered for patients with elevated Lp(a) and progressive cardiovascular disease and is the only FDA-approved therapy to lower Lp(a) in the US. LA reduces Lp(a) by 70% in patients with elevated Lp(a) and cardiovascular disease.79 Although no randomised controlled studies have been conducted to demonstrate the effectiveness of LA in reducing cardiovascular events, LA has been shown to be associated with a 94% reduction in cardiovascular events over a mean treatment period of 48 months.79 In the Pro(a)LiFe study, patients with high Lp(a) were prospectively followed, and the incidence rates of cardiovascular events between 2 years before and 2 years after LA treatment were compared. The mean rate of cardiovascular events declined from 0.41 for 2 years before LA to 0.09 for 2 years during LA.80 LA effectively lowered the incidence of cardiovascular events with a significant reduction in Lp(a). Both the German and UK apheresis guidelines use Lp(a) >1.55 mmol/l (60 mg/dl) to specifically allow reimbursement for patients with isolated Lp(a) elevation and recurrent cardiovascular events or in conjunction with uncontrolled elevated LDL cholesterol.81,82
Conclusion
Several scientific approaches, including observational studies, genetic studies, and Mendelian randomisation studies, have clarified the clinical impact of elevated Lp(a). A level of 1.29 mmol/l (50 mg/dl) has been suggested as a practical cut-off point for risk stratification for future cardiovascular events in daily practice, while a level of 0.78 mmol/l (30 mg/dl) may be an alternative cut-off for Asian populations. In addition to people with a high risk of atherosclerotic cardiovascular disease, the healthy general population may also be a potential candidate for Lp(a) measurement. Although the optimal pharmacological intervention for elevated Lp(a) has not been established, some ongoing trials may open the door to novel treatment strategies for people with elevated Lp(a). Healthcare providers must pay more attention to the risk stratification of cardiovascular disease according to Lp(a) level.
Clinical Perspective
- Elevated lipoprotein (a) (Lp(a)) is associated with an increased risk of cardiovascular disease.
- Plasma Lp(a) >1.29 mmol/l (50 mg/dl) has been proposed as a practical cut-off.
- An alternative cut-off of Lp(a) for Asian populations may be 0.78 mmol/l (30 mg/dl).
- Measurements may have to be considered at least once in the lifetime of each adult.
- Proprotein convertase subtilisin kexin type 9 inhibitors and antisense oligonucleotides may reduce the Lp(a) plasma level.