









It is well established that ageing is a preordained part of life, which crosses gradual deteriorations in physiological functions with an elevated risk of health complications and chronic diseases. Among all, the impact of ageing is a crucial predictive factor of one’s cardiovascular health.1 It was projected that by year 2030, 40% of all deaths across the population group aged ≥65 years will constitute cardiovascular diseases (CVD). Age-associated heart morphological and functional changes predispose to cardiac vulnerability with ageing.1–3 Yet, the underlying descriptions and molecular genetic mechanisms of age-related vascular changes remain poorly understood.
Telomeres are short tandem repeat DNA sequences, (TTAGGG)n, located at the ends of eukaryotic chromosomes. These repetitive codes are non-coding DNA regions that shield the degradations of nearby genes during cell divisions and senescence. Although telomere length (TL) varies significantly among individuals, it progressively shortens with age and each cell cycle, and has been recognised as an ageing biomarker.4 Recent meta-analyses have established the association of TL, as proposed in leukocyte TL (LTL), with cardiovascular disease and its risk factors.5,6 Yet, this biological association remains ambiguous, as shown by indolent inflammation and oxidative stress.7,8
The reduced and/or compromised functions of myocardial cells or vessels accounts for the acute and chronic onsets of myocardial dysfunctions. Both systems establish a dynamism, affecting each other through compensatory mechanisms where chronological age appears as a predictive factor (but not as a causal factor). In fact, it reflects on the progressive and accumulative effects of ageing on endothelial damage in times of various mechanical, haemodynamic and immunological impacts.9 Indeed, age-related telomere attrition (or telomere shortening) and CVD have a substantial relationship.10 This is likely due in part to the associations of TL with multiple CVD risk factors. Epidemiologically, unstable atherosclerotic plaques presented with shortened LTL and low telomerase activity, which corresponded to increased risks of stroke and MI.11 In addition, myocardial tissue regenerative capacity is compromised at the time of ageing myocardium with LTL attrition and extensive cell senescence, thus being conducive to systolic/diastolic heart failure.
Independent of age, the rate of telomere shortening is also highly influenced by oxidative stress and inflammation, both of which are intricately significant drivers of CVD. Patients with ion channel disorder showed genomic imbalances in key ion channel genes and oxidative stress-associated telomere attritions, which may give rise to sudden cardiac death.12 Chronic oxidative stress in a cardiac microenvironment driven by a defective ion channel might expedite the telomere attritions in both heart tissues and peripheral leukocyte DNA, which would possibly support the partial causes of sudden cardiac death in young people.
At this juncture, it remains elusive whether telomere attrition demonstrates a mitotic timepiece process or, rather, if it is a stress biomarker or a biological proxy taking charge of life-course stress exposures and stress-associated signalling mechanisms to the cell.13–15 Despite the uncertainty, the accumulated hypotheses support the involvements of telomere attrition and telomerase activity in the onset, development and mortality of CVD. Telomere length remains as one of the putative biomarkers for risk stratification of CVD, and is being increasingly used as a promising biomarker in personalised medicine.16 In this brief review, we provide a concise update, and diverging view on telomeres, telomerase and their relationship to CVD. In addition, we discuss the open issues and challenges of current diagnostic and therapeutic interventions centred on the telomere biology in CVD.
Telomere and Telomerase Biology
The repetitive TTAGGG DNA sequences of telomeres cap the chromosomal ends to retain genomic integrity.17 During each mitotic cycle, 20–300 nucleotides are lost at the 3´-hydroxyl end of DNA through incomplete synthesis of the lagging strand – commonly referred to as the ‘end replication problem’ phenomenon.18–19 Telomeres are protected by six capping proteins, otherwise also known as the shelterin complex (Figure 1). These proteins are arranged into a T-loop structure to prevent the telomeres from being mistakenly recognised by the DNA damage sensors for DNA breaks.20 Hence, TL is proposed as a mitotic timepiece to assess the number of repeated cell divisions.
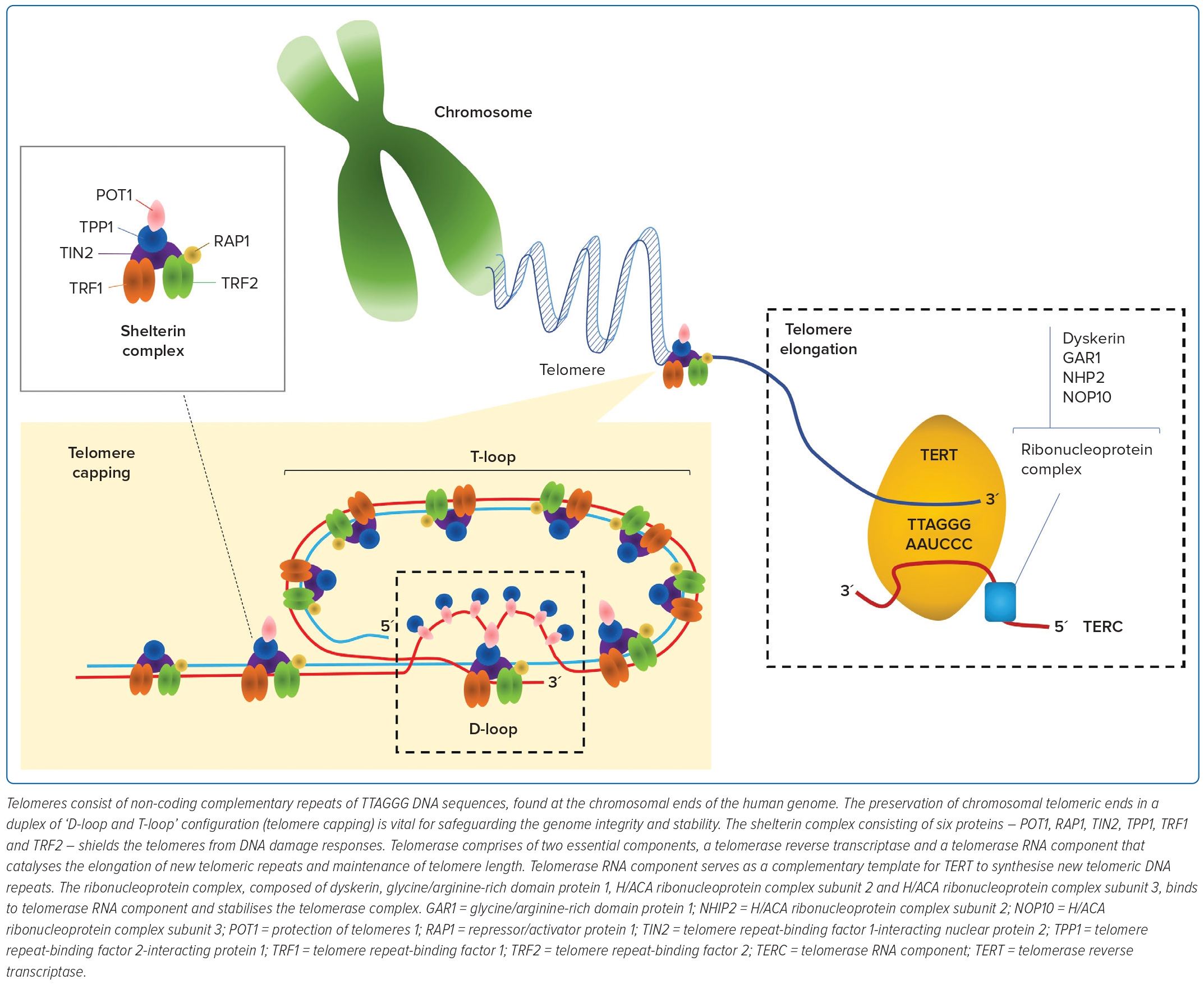
The maintenance and elongation of TL are regulated by telomerase. To maintain TL, telomerase uses telomerase RNA component and telomerase reverse transcriptase (TERT) to catalyse the synthesis of a new telomeric DNA sequence at the single-stranded overhang. High-level telomerase activity is found in most cancer cells, activated lymphocytes, stem cells and haematopoietic progenitor cells to prevent TL attrition, and preserve infinite cell division. In contrast, telomerase activity in somatic cells is generally at a low-to-undetectable level and has a constrained longevity.
Although the loss of telomerase activity is accompanied by physiological ageing, it markedly increases in response to injuries.21 For instance, the telomerase expression in the mammalian heart is naturally low, yet functionally significant. Cryoinjured adult mice hearts showed significant upregulations in telomerase expressions within the cardiomyocytes, endothelial cells and fibroblasts, suggesting the tissue repair and regeneration roles of telomerase.22
TL is determined by genetic and non-genetic factors. Cells naturally enter replicative senescence when the shortened TL approaches a critical length incompatible with its functioning, thus resulting in cell cycle arrest and triggering the apoptotic pathway driven by the gene expressions of replicative cell cycle inhibitors.23 Separately, cell senescence can be triggered by external stressors, a process known as stress-induced premature senescence.
Oxidising agents and radiation are common external stimuli that drive the premature activation of cell senescence independently of telomere attrition. Under the oxidative stress environment, senescent cells undergo morphological changes, and secrete high levels of inflammatory cytokines and immunomodulators in autocrine and paracrine patterns – a process known as senescence-associated secretory phenotype.
Commonly secreted inflammatory cytokines and immunomodulators include interleukin-6, interleukin-8, vascular endothelial growth factor and plasminogen activator inhibitor-1, which drive a vicious cycle of inflammation, apoptosis, tissue remodelling and repair.24,25 The G-rich telomere repeat sequence poses a unique feature that predisposes telomeres to oxidative damage, thereby driving the cell into senescence.26 Hence, cumulative exposure of oxidative stress and inflammation can aggravate chronic inflammation, and accelerate ageing and the progression of ageing-related disorders.25
Genetic and environmental factors can also influence the interindividual variations in TLs across different tissues.27,28 TLs are likely synchronous at birth (within an individual, but remain variable across different entities), with good correlations among various cell and tissue types. However, variations mostly take place during extrauterine life.29 For instance, muscle tissues and the heart in theory would undergo different proliferation rates, as compared with blood and skin cells. TLs and their shortenings reflect inheritable genomic features, past replication history and accumulative exposures to non-genetic factors. As such, TLs tend to be highly asynchronous across different cell and tissue types after birth, and their respective shortenings likely occur at a different rate. Although, the biology of age-associated TL shortening is extremely complex and the biological mechanisms underlying this process are yet to be definitively established. Nonetheless, TLs generally shorten with premature (due to external stressors independent of age) and chronological ageing in all proliferating tissues, and they share common manifestations of reduced tissue regenerative capacity, escalated inflammatory responses and, eventually, prominent organ dysfunctions.30,31
Association of Telomere and Telomerase with CVD
The associations of leukocyte TLs and telomerase activities with atherosclerotic and cardiovascular risk factors are well reported in the literature.4,32 The risk factors, such as ageing, hypertension, diabetes, obesity, sedentary lifestyle and smoking, have been associated with short LTL. Among these, ageing is considered a major risk factor for CVD, which includes MI, stroke, hypertension and heart failure. The main mechanisms underlying CVD are oxidative stress and inflammation, which have been reported to accelerate telomere shortening and eventually lead to cellular senescence. Hence, a better understanding of the interaction between ageing, oxidative stress and the telomere–telomerase system is required for the development of therapeutic interventions for CVD treatments.
Fundamental Research: Animal and Human Cells
In Vitro Studies of Telomeres and Telomerase
Acceleration in telomere shortening with increasing age is considered to be the typical characteristic of molecular ageing.33 Short telomeres are associated with cardiac dysfunction in mice and humans with CVD.34,35 A lack of TERT activities has been associated with the development of cardiovascular dysfunction.36 Overexpression of TERT in mouse cardiac muscle promotes telomerase activities and telomere lengthening, and extends the division capacity of cardiomyocytes.37 Telomerase activation could induce telomere lengthening and confer cardioprotection in adult mouse heart after acute MI.38 Downstream alterations due to telomere shortening have been reported to induce cell cycle arrest in cardiomyocyte and cellular senescence.36 Telomere-induced senescence and apoptosis have been demonstrated through the expression of p53 and p16, which are the common molecular attributes associated with a reduction of cardiomyocytes. The decreasing proliferative potential of cardiomyocytes also restricts the regenerative capacity of aged and injured myocardium and vasculature.39,40
In a study conducted on telomerase knockout mice, telomere shortening and telomerase deficiency resulted in impaired cardiomyocytes regeneration. This leads to pathological cardiac remodelling and severe ventricular dysfunction in late-generation telomerase RNA component-deficient mice.36 Age-associated phenotypes could be reversed in a telomerase-deficient model of accelerated ageing via reactivation of telomerase. In addition, stimulation of telomerase could result in favourable metabolic improvements in adult organisms without cancer.41
A study also showed suppression of telomere attrition and prevention of terminal differentiation of cardiomyocytes in the first month of life in mice myocardium subjected to TERT overexpression.37 An increase in the median lifespan of 24% and 13% was reported in 1- and 2-year-old telomerase-treated mice, respectively, but not with catalytically inactive TERT. This demonstrates that the essential role of TERT and the potential of telomerase-based therapy in prolonging the lifespan of normal mice.42 In primary human cells, transfer of an exogenous human TERT complementary DNA enables the prevention of telomere shortening and immortalisation of primary human cells without cancer-related alterations.43
When telomeres shorten with age, somatic mutations in recurrent genes in haematopoietic stem cells leads to an age-dependent accumulation of mutated blood cell clones called clonal haematopoiesis of indeterminate potential (CHIP).44,45 Telomere attrition and CHIP are associated with age.46 Clonal haematopoiesis is considered as an emerging cardiovascular risk factor associated with the development of atherosclerosis and cardiac dysfunction.45 A CHIP–atherosclerotic association has been studied in a murine model with loss-of-function mutation in TET2. In the LDL receptor knockout mice prone to atherosclerosis, activation of NLRP3-mediated inflammasome and accelerated atherosclerosis were observed after receiving bone marrow from TET2-knockout mice.47,48
CHIP mutations in mouse haematopoietic cells have been shown to exhibit the development of heart failure. In the same study, haematopoietic cells with TET2 deficiency was also associated with greater cardiac dysfunction resulting from interleukin-1β signalling elevation.49 The presence of CHIP in peripheral blood cells has increased cardiovascular mortalities independently of traditional risk factors. In addition, the presence of CHIP in peripheral blood cells was also associated with an increased risk of coronary heart disease in humans and with accelerated atherosclerosis in mice.47
Clinical Research: Humans
Clinical Studies of Telomeres and Telomerase
CVDs have been inadvertently associated with the inherent percentage of short telomeres. TLs were found to be significantly shorter than that in controls, and were inversely correlated with the severity of this disease in patients with coronary heart disease.50 Differences in biological ageing could affect susceptibility to coronary heart disease among individuals.51 A comprehensive review showed the importance of telomeres on cardiovascular ageing, and concluded that an increasing telomere attrition rate due to cardiovascular ageing-related factors could be complemented by telomere-mediated haematopoietic senescence.34
Increasing evidence shows that critical telomere shortening has a crucial role in cardiac ageing and disease. The implication of short telomeres on heart function and the roles of telomerase in mitochondrial modulation demonstrate that telomere attrition, senescence and cell death are associated with the development of CVD in humans.52
Increasing senescent cardiomyocytes, shown by the expression of senescence markers p53, p21 and p16, and the presence of shorter telomeres, were reported in ageing hearts.39 In the aged heart, critical alterations occur, such as functional loss of cardiomyocytes and other supporting cells, due to clearance of dysfunction cells.2,53 The loss of cells also leads to the reduction of heart regenerative capacity as ageing progresses.54 Telomere dysfunction can induce p53-dependent repression of the main regulators of mitochondrial biogenesis and function, peroxisome proliferator-activated receptor gamma coactivator-1α, and peroxisome proliferator-activated receptor gamma coactivator-1β in the heart. This leads to bioenergetic compromise due to reduced oxidative phosphorylation and adenosine triphosphate generation. Hence, factors associated with heart ageing, such as p53 activation and mitochondrial dysfunction, contribute the molecular basis of dysfunctional telomeres in affected cardiomyocytes during the progression of cardiac ageing.52
Atherosclerosis is an inflammatory disease characterised by immunological activity that involves vascular endothelium, smooth muscle and blood cells.55 It is considered as the prevalent pathology of CVD, including MI, heart failure, stroke, hypertension and peripheral artery diseases.56 Short telomeres are indicative of a higher risk for atherosclerosis and related vascular complications.57,58 It has been reported that short LTL and low telomerase activity were associated with instability of atherosclerotic plaque, and a higher risk for coronary heart disease and MI.59–61 Besides blood, shorter telomere lengths were also detected in vascular tissues from patients with atherosclerotic disease, such as aortic tissues, which showed atherosclerotic lesions.62
Short LTL has been associated with a heightened likelihood of developing hypertension. The shortening of telomeres plays a role in causing endothelial dysfunction, stiffness in arteries and inflammation − all factors connected to elevated blood pressure and the pathological processes in CVD.63 In patients with atherosclerosis, shorter telomeres were present in coronary endothelial cells, compared with health individuals. Vascular endothelial cells with senescence-associated phenotypes are observed in human atherosclerotic lesions with altered functions via inhibition of telomere repeat-binding factor (TRF)-2 alone. This suggests that telomere shortening induces endothelial cell senescence, and telomere function is necessary for endothelial function.64
In atherogenesis, short telomeres in endothelial cells might increase pro-inflammatory reactions and promote unstable atherosclerotic plaques. Study on glutathione-dependent redox homeostasis demonstrates that preservation of telomere function from oxidative stress-induced premature senescence of endothelial cells might contribute to pathogenesis of vascular disease.65
Arrhythmias and heart dysfunction due to a lack of regenerative capacity in aged cardiomyocytes and heart stem cells has been associated with short telomeres and a high risk of cardiovascular disease in healthy humans.34,52 Shorter telomeres may contribute to cellular dysfunction and oxidative stress in cardiac tissues.66 Aged hearts have an increased number of senescent cells with expression of p16, p21 and p53, as well as the presence of short telomeres, which might contribute to the development of cardiac failure.39,67 Biopsied tissue from a failing heart showed the presence of shorter telomeres and DNA damage specific to cardiomyocytes, regardless of the patient’s age.68
In a meta-analysis of 14 genome-wide association studies consisting of 22,233 cardiovascular disease patients and 64,762 controls, seven single-nucleotide polymorphisms were identified for the variation in mean LTL. It was observed that a mean of 117 base pairs of telomere length shortening per telomerase RNA component telomere-shortening allele contributes to an almost 10% decrease in telomere reserves in middle-aged adults. This also promotes the susceptibility of individuals to replicative senescence and dysfunctional telomeres.69
In a study conducted on longitudinal changes in LTL in relation to all-cause, cardiovascular and cancer mortality in 247 elderly Swedish men, short baseline telomeres were linked to mortality risks, with 40−70% increased risk of all-cause mortality. Longitudinal changes in telomere length showed shortening in 83% of individuals, and the study reported an increased risk of mortality in individuals with short baseline telomeres without any relationships to all-cause and cancer mortality for changes in telomere length.70
Pusceddu et al. investigated the relative telomere length in 3,316 participants of the LURIC study, and reported that patients in quartiles 2 ± 4 of relative telomere length had a lower hazard ratio for all-cause mortality and CVD mortality when compared with those in the first quartile.71 This study also demonstrated an 18% risk reduction in all-cause mortality and 16% reduction in cardiovascular disease mortality in patients with the longer telomeres when compared with those with the shortest telomeres, suggesting that short TL increases the risk of all-cause and cardiovascular mortality. An increase in all-cause mortality in studied individuals with short telomeres was also reported in the CHS and the LIFE study for high-risk populations.11,72
In the WOSCOPS study, TLs in 484 individuals who developed coronary heart disease were compared with 1,058 matched controls. It was found that individuals in the lowest tertile of leukocytes TL had a 44% increased risk of coronary artery events, as compared with individuals in the highest tertile in a mean follow-up period of 5.5 years after adjustment for risk factors for coronary artery disease.51
The rate of changes in LTL was examined in 236 randomly selected white participants from the MacArthur Health Ageing Study aged 70−79 years. The result showed that short baseline LTL was linked with 2.3-fold higher risk of mortality in CVD in women, whereas age-related LTL shortening, but not baseline LTL, was linked with a threefold higher risk for cardiovascular mortality.73 Hence, differences in biological ageing between individuals could also associated with the susceptibility to coronary heart disease.
In patients with premature acute MI (aged <50 years), LTL was found to be shorter than in healthy and age-matched controls.74 It was estimated that each kilobase pair shortening of telomeres in peripheral blood cells resulted in 2.8- to 3.2-fold higher risk of MI and stroke.75 Leukocyte telomere shortening was also reported to be significantly linked to cardiovascular outcomes in patients with ischaemic heart failure.76 A decrease by approximately 40% of LTL in patients with heart failure was found in a study that involved 803 patients.77
In contrast, shorter LTL has been associated with an increased risk of developing peripheral artery disease (PAD), suggesting that telomere length may be a potential biomarker for the susceptibility to PAD. A cross-sectional study reported that the LTL was associated with PAD, and the SD decrease in the length significantly increased the risk of PAD by 44%.78
In a prospective study for a follow-up period of 6 years involving 768 patients, telomere shortening was associated with increased carotid artery intima-media thickness and an increased rate of cardiovascular cases after CVD risk factors adjustment.79 In addition, LTL may influence outcomes following revascularisation procedures (e.g. angioplasty, stenting, bypass surgery). Patients with shorter telomeres may have a higher risk of adverse events or restenosis after such procedures. Yin et al. reported that asynchronous shortening of the telomere length between leukocytes and cardiac atrial tissue can serve as a better biomarker than leukocyte length alone for postcardiovascular surgery events.80 They discovered that the extent of the difference in telomere length between cardiac atrial tissue and leukocytes was linked to both postoperative complications and the duration of stay in the intensive care unit after surgery.
Methodological Aspects
Although TL is conceivably asynchronous across different tissues, peripheral blood LTL remains commonly assessed in transverse and longitudinal clinical studies owing to: its minimal invasiveness, hence permitting safe and repetitive samplings on test subjects, and its adoptability on in vivo animal models.11,28,81 Multiple techniques, such as TRF analysis, single telomere analysis, flow cytometry- or digital microscopy quantitative in situ hybridisation and quantitative PCR have been developed to measure TL.82 Unfortunately, there is still no current gold standard of TL measurement, and this has hampered its wider application.
Although a single shortest telomere is capable of triggering the cellular senescence process, the overreliance on measuring the average TL remains a key limitation of current approaches, which lack representativeness to associating TL with the complex biology of ageing.83 Nevertheless, each of these developed TL methods remain useful, and their respective merits and limitations are summarised in Table 1.84–96

All in all, considering the merits and limitations of different technological approaches, the substantial interlaboratory variations and the discrepancies across intra-assay results could still mislead the final result interpretations of relating TL to ageing and disease pathology. Aside from the discrepancies in assay principles and procedures, other determinative factors, including sampling methods, preparation, processing and storage, should also be considered to ensure the accuracy and reproducibility of TL measurements.90,91 Hence, additional optimisations of currently available techniques remain warranted to improve the sensitivity, reproducibility and correlativity of their respective outputs.
Potential Treatment Interventions
Many studies have reported that maintenance of telomere length and modulation of telomerase activity reverse telomere attrition and cellular senescence.37,41 These findings also serve as potential therapeutic interventions and emerging novel strategies for treatments of atherosclerosis and cardiovascular disease.97 Hence, prevention of senescence in young individuals and restoration of cardiovascular functions in older adults could be the future goal of telomere cardiovascular therapy.98
It has been reported that the use of modified messenger RNA for in vitro synthesis of TERT in human fibroblasts was able to increase telomerase activity, the length of telomeres and proliferative capacity.99 This was achieved when mouse TERT with an adeno-associated virus was transferred into young and old mice via telomerase gene therapy, resulting in elongated telomeres and extended lifespans.35 In a preclinical mouse model treated with TERT gene therapy after acute MI, improvement in ventricular function and limitation of infarct scars were observed.38 Hence, TERT gene therapy could be a promising technique that deserves further investigation for the treatment of age-related diseases, such as CVD.
Pharmaceutical intervention, such as prescribed medication for treatment of CVD, has been reported with effects of telomere length maintenance and senescence prevention. Statins, also known as 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors, are used to prevent the development of atherosclerotic plaque. Their activities include enhancement of DNA repair capacity and upregulation of glutathione synthesis to combat oxidative stress.100,101 Statins have been reported to enhance telomerase activity and protect telomeres through upregulation of the TRF-2 in endothelial cells and endothelial progenitor cells.102,103 In a cross-sectional analysis of 3,496 subjects from the US National Health and Nutrition Examination Survey, longer telomere length was observed with a longer duration of statin usage.104 Higher telomerase activity independent of other covariates, such as age, smoking and lipid profile, was associated with statin treatment.105
Angiotensin II has been reported to induce DNA damage caused by oxidative stress, and promote senescence in cultured human vascular smooth muscle cells.106 Long-term exposure of vascular smooth muscle cells to angiotensin II has resulted in the reduction of proliferation and replicative senescence with telomere shortening. Hence, inhibitors of angiotensin II or angiotensin-converting enzyme can be used as potential medication to reduce oxidative stress and cellular senescence.107
Both angiotensin II receptor blockers, such as losartan, and angiotensin-converting enzyme inhibitors, such as captopril, have been reported to protect endothelial progenitor cells from senescence and dysfunction.108,109 Angiotensin-converting enzyme inhibitors have anti-oxidant properties that exert upregulation of endothelial nitric oxide synthase, fibroblast growth factor-2 and TERT messenger RNA. This also contributed to promoting endothelial cell survival and restoration of endothelial cell functions after vascular damage.109
Pioglitazone, a peroxisome proliferator-activated receptor agonist, has been reported to increase the activity of telomerase and expression of TRF-2 in mice aorta. In addition, reduced senescence markers, p16, cell-cycle checkpoint kinase 2 and p53 were observed in pioglitazone-treated mice.110 Increased TERT and TRF-2 expression in the hearts of diabetic rats after treatment with pioglitazone has also been reported.111 Pioglitazone can also reverse angiotensin II-induced endothelial progenitor cells senescence through telomerase activity enhancement.108
A bioactive molecule extracted from Astragalus membranaceus, TA-65, is the first telomere activator to be described. It has been used in Chinese traditional medicine as an anti-ageing drug, and is reported to have effects on telomere lengthening in mice.112 Treatment with TA-65 has been shown to induce telomerase-dependent elongation of short telomeres and reverse DNA damage in fibroblasts. In addition, dietary supplementation of TA-65 also showed improvement of several health indicators in the cardiovascular system and metabolism in a human study.113 In a randomised, double-blind and placebo controlled clinical trial, TA-65 treatment was shown to increase LDL cholesterol and reduce C-reactive protein in patients with metabolic syndrome, and was also found to elongate telomeres.114,115
Open Issues and Challenges
Peripheral leukocyte DNA is commonly used in clinical cardiovascular health studies to assess LTL.11 Although shorter, LTL has demonstrated a significant association with cardiovascular adversity. Its causal role in the epidemiology of CVD remains controversial, and is attributable to several limitations of TL in clinical studies.
First, the distinction in study designs is hindering the interpretations of TL in CVD. It should be acknowledged that the design of longitudinal research (prospective or retrospective study), assay application methods, patient characteristics and the relative risks of different disease outcomes are some of the key factors that need to be considered before generalising the study results.5
Second, the correlation of TL in different tissues to CVD remains unknown. Previous human studies on telomere attrition rates in leukocytes, subcutaneous fat, skin and skeletal muscle have demonstrated strong correlations across these four tissues.116 Most clinical studies measure LTL based on the assumption that it is a representative of TLs in other tissues or organs. In retrospect, TL in normal somatic tissues of other mammal species (dogs and pigs) have demonstrated extensive heterogeneity.117,118
Third, due to the low leukocyte telomere attrition rate, most observational studies on time-dependent (age) changes in TL were conducted by comparing patients of different age groups. Such study design tends to overlook the complex dynamics of TL and its heterogeneity across different individuals in most of the cross-sectional study.
Fourth, although TL is offered as a solid biomarker for biological ageing, sepsis, inflammation, viral infection and environmental stresses can directly affect the rate of telomere attrition independent of ageing.119–122 These factors can predispose a cardiovascular-associated disease outcome. Considering all these limitations, the strengths of LTL to be offered as a solid clinically relevant cardiovascular risk marker remain to be explored further.
LTL varies significantly among individuals from birth onwards, and there is a wide LTL variation across newborns, challenging the telomeric clock model.123 An individual’s LTL is influenced by dynamic factors, such as LTL at birth.124 Consequently, whether an individual has a short or long LTL may be determined at birth. Understanding the significance of this initial setting in later life, particularly regarding disease manifestation in adulthood, is crucial.123
Martens et al. conducted a study on tracking TL in newborns, and observed changes from birth to childhood and adulthood in two birth cohorts. The results indicated that TL at birth can potentially predict TL later in life.125 Furthermore, a higher attrition rate of TL was noted in individuals with longer TL at birth. The study also revealed that a longer maternal TL is associated with a lower telomere attrition rate in the next generation. However, tracing back TL at birth for each study subject remains a challenge, considering the dynamics of defining telomere dynamics.
It is important to note that lifestyle habits can also influence TL attrition rate. Comprehensive lifestyle habits, such as aerobic exercise, have been shown to slow the TL attrition rate. Aerobic exercise is generally known to have beneficial preventive effects against vascular diseases due to the reduction in oxidative stress.126 The TL attrition rate was found to be reduced with improved physiological anti-oxidant activity and redox balance resulting from regular exercise.127 Additionally, athletes exhibited a lower telomere attrition rate and higher telomerase activity compared with the inactive control study pool.128 A recent study by Song et al. also reported that aerobic exercise for >6 months shows positive improvement in telomere length.129 Therefore, apart from traditional TL-associated risk factors, other age-independent factors (such as comprehensive lifestyle habits) of study subjects should be considered as adjuncts to TL. This consideration is essential to support TL dynamics as a reference biological proxy in the manifestation of CVD.
The process of biological ageing is far more complex than chronological ageing. Early human studies proposed that TL and the rates of telomere attritions were tissue independent.116,130 However, it was recognised that the small sample sizes and the difficulties in acquiring different tissue samples affected the rationale of the original inference. In addition, the present TL measurements consider large and different cell populations to represent the overall average TL, while the heterogeneity of single-cell telomere dynamics is often excluded from the context. Separate findings from another research group later demonstrated the different rates of telomere shortening across different tissue.80 In human hearts, the rate of telomere shortening in cardiomyocytes is different from other cell types.28 Moreover, due to the slow nature of the telomere attrition rate, the sample sizes and study durations can affect the overall study statistical power and interpretation of results.
In theory, different tissues may share an identical process of biological ageing at baseline. However, some parts of the physiological systems would exhibit different telomere attrition rates, as driven by accelerated biological ageing or age-independent factors before the TL reaches a critical length of systemic failure. Hence, having a good autologous referencing approach would be pertinent to delineate telomere dynamics in the process of biological ageing and to gauge the extensiveness of telomere attrition over time. In addition, studies focusing on cell type-specific telomere shortening via a single-cell telomere length assessment approach could be incorporated to expand the horizons of telomere dynamics in CVD, and possibly to use the collected indices in raising therapeutic goals and outcomes.80
Telomerase stabilises the critically short telomeres by elongating the chromosome ends de novo.131 The enzyme is actively expressed in the early foetal development (in utero) stage, in proliferating stem cells of highly regenerative tissues (such as germ line cells), embryonic stem cells and immune cells.132 Although telomerase remains expressed in the stem cell compartments, it still fails to cope with maintaining normal TL; gradual telomere shortening still occurs in these cells.85,133 In the somatic cell compartment, telomerase activity is low-to-none in most normal somatic tissues and remains heterogenous across different cells.134 Cardiomyocytes, endothelial cells and putative cardiac stem cells in murine hearts all exhibited heterogenous telomerase activities.135 Nonetheless, the homeostasis of cardiac telomeres is likely regulated by the low-level telomerase activities of these small cell populations.38 Hence, considering the biological significance of telomerase in the maintenance of TL, telomerase activators have been considered as a promising anti-ageing therapeutic option for degenerative disorders.136,137 In contrast, telomerase is reactivated in most human malignancies, and this has cast doubts on the clinical applicability of telomerase activator.138
Conclusion
Although in vitro and in vivo evidence of telomere and CVD is convincing, they are still far too inconclusive to be translated into clinical use. It remains debatable whether to focus on TL and/or telomerase as prospective diagnostic and/or therapeutic target(s) of CVD. Differences in assessment methods of TL, study designs, the lack of gold reference standards and many other factors contribute to the resultant inconsistency and discrepancy of outcomes across the reported studies. This has led to debates on the reliability of TL as a valid reference to assess age-associated TL attrition. In addition, the biological mechanisms underlying age-associated telomere attrition remain complex, and have yet to be conclusively defined. It remains unclear whether telomere attrition reflects a chronological ageing process, a stress biomarker or a signalling mechanism that drives stresses to cells and tissues. In the event of a stress-related cause, telomere attrition could represent an intermediary of lifetime stress exposure.
Considering these uncertainties, the associations and impacts of telomeres, telomerase, and TL on the aetiology of cardiovascular disease are still not definitively established. Nonetheless, it is generally agreed that the rate of telomere attrition is plausibly related to atherosclerotic development, and it remains as one of the commonly used cardiovascular disease mortality predictors in patients. Until the known open issues and challenges of telomere and telomerase are definitively addressed, it is worth noting that at the present time, additional risk factors and biomarkers (in addition to elements of telomeres) remain mandatory for diagnosing and monitoring the therapeutic interventions of CVD. 
Clinical Perspective
- Telomere length (TL) may provide insights into the progression and severity of cardiovascular disease (CVD). Individuals with shorter TL may experience a faster progression of CVD, increasing their susceptibility to adverse cardiovascular events.
- Variations in the evaluation techniques of TL, diverse study designs and the absence of universally recognised gold standard references were some of the consideration factors towards the credibility of TL as a single, reliable indicator for assessing age-related telomere attrition in CVD.
- It remains essential to emphasise that, presently, the inclusion of additional risk factors and biomarkers, alongside aspects of telomere biology, is imperative for the diagnosis and effective monitoring of therapeutic interventions in CVD.
- Therapeutic interventions aimed at preserving or lengthening telomeres – via telomerase activation – could be explored to mitigate the progression of CVD. In addition, stratifying patients based on TL for tailored treatment strategies may lead to more effective disease management.