









Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), arising in Wuhan in autumn 2019, has severely affected both economies and health worldwide over the past 2 years. Although most infected individuals are asymptomatic, 10–15% of cases foster critical conditions featured by pneumonitis or multiple organ failure.1 The published mortality rate for critically ill patients requiring mechanical ventilation is persistently high, and the recuperation period for these patients is prolonged.2 The scale of long-term restoration of integrity is not known in every respect yet. However, clinical data reveal that unimpaired recovery is often not obtainable, particularly in individuals with associated viral myocarditis or multiorgan involvement.3 Numerous diverse therapeutic interventions have been proposed for viral pneumonitis with some degree of success, but a specific treatment has not yet been established. Ongoing vaccination programs limit disease spread and the extent of disease severity in affected patients.4 This review analyses the role of COVID-19 and the pathomechanisms underlying its short- and long-term cardiac effects.
Search Strategy and Selection Criteria
Appropriate data were identified by searching the PubMed database, as well as reference lists in relevant articles using the search terms ‘SARS’, ‘COVID-19’, ‘renin–angiotensin system’, ‘phagocyte’, ‘reactive free radical’, ‘antioxidant’, ‘ARDS’, ‘thrombosis’, ‘acute myocardial’, ‘ischaemia’, ‘reperfusion’, ‘microvascular’, ‘ACE2’, ‘heart failure’, ‘haemodynamics’, ‘cardiac markers’, ‘myocarditis’, ‘cardiomyopathy’, ‘transplantation’, ‘ECMO’, ‘ventricular assist device’, ‘vaccine’ and ‘medical therapy’. Abstracts and meeting reports were not included in the present study. Only articles published in English between 1976 and 2021 were reviewed.
This paper is a scoping review; therefore, a detailed risk of bias assessment was not conducted. The data were abstracted by investigator one (LG) and verified by the second investigator (YM); discrepancies were resolved by discussion. Final inclusion was determined by the senior investigator (EMT).
COVID-19 Development and Angiotensin-converting Enzyme 2
Angiotensin-converting enzyme (ACE) 2 is a transmembrane peptidase. It is the receptor for the Severe Acute Respiratory Syndrome (SARS) coronavirus that caused epidemics in Guangdong, China, and Canada in 2003.5 ACE2 is expressed in numerous organs, including the intestinal epithelium, heart, central nervous system and kidneys, with high density in the microvascular endothelium. SARS-CoV-2 targets the ACE2 isoform located on the surface of Type I and II alveolar pneumocytes during initial respiratory infection.6 In addition, ACE2 expression is observed on alveolar phagocytes and lymphocytes, having a pivotal role in the leucocyte activation process.7
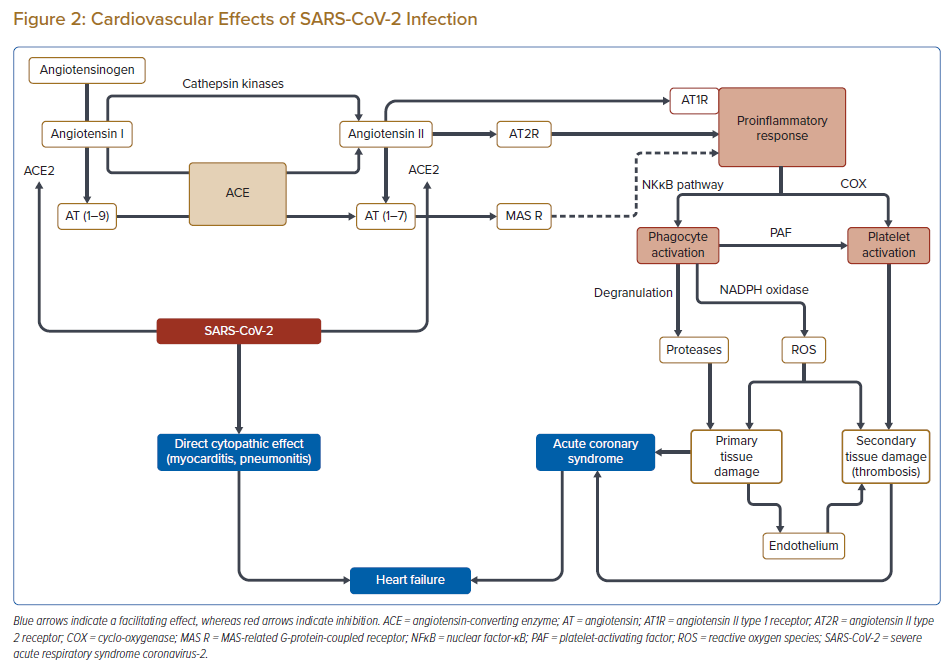
ACE2 splits angiotensin I (ATI), into angiotensin (1–9), thereby inhibiting the renin–angiotensin system (RAS). In addition, ACE2 transforms angiotensin II (ATII) into angiotensin (1–7), with angiotensin (1–7) activating the MAS-related G-protein-coupled receptor (MAS R), resulting in an anti-inflammatory effect and vasodilation.8–11 The coronavirus connects via its spike protein (S-protein) to ACE2 outside the enzyme’s catalytic domain, triggering virus internalisation into the cell expressing the receptor and viral replication.8,9,12
In SARS and Middle East Respiratory Syndrome, mortality ensued not from a direct cytopathic effect of the coronavirus, but rather acute respiratory distress syndrome (ARDS), a culmination of maladaptive and excess immune responses of the host, generated by RAS dysfunction leading to reduced ACE2 activity.13,14 As a result of the RAS imbalance (ATI-angiotensin 1–7), the elevated ATII level triggers a pathologic inflammatory response by the angiotensin II type 1 receptor (AT1R). Activation of AT1R initiates the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) signalling pathway, resulting in a surge in tumour necrosis factor-α (TNF-α) and interleukin IL-1 and IL-6, activating the circulating neutrophil (PMN) cells and monocytes/macrophages (MPH). This activation causes further adhesion and migration of phagocytes by facilitating the expression of Vascular cell adhesion molecule-1, monocyte chemoattractant protein-1 and intercellular adhesion molecule-1.15–19 The vascular marginalisation, endothelial rolling, diapedesis and tissue migration of phagocytes leads to an oxidative burst (i.e. exaggerated production of free radicals/reactive oxygen species; Figures 1, 2).20,21 The expression of novel viral proteins (e.g. ORF3b, ORF6, nucleoporin Nsp1, papain-like protease) intensely influences the innate immunity on numerous targets. Hindered production of critical antiviral interferons has been demonstrated in human cell cultures and murine models, and amplified synthesis of cytokines (IL-6, IL-8) regulated by NFκB signalling has been reported.16 The latter may play a critical role in the pathogenesis of ARDS.22–24
Phagocyte Actions in Host Tissue Damage
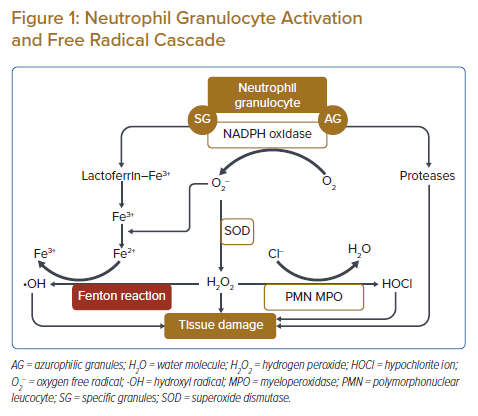
Circulating PMNs use reactive oxygen species to defend the body against invading germs and foreign particles.25 However, these defensive pathways can also pose a risk for the host; in the case of an imbalanced enzymatic or oxidative response to a harmful substance, there may be damage caused to host tissues.26 After leaving the bone marrow, PMNs travel to the capillaries and then reach post-capillary high endothelial venules (HEV); diapedesis starts through the intercellular junctions. Over a 5- to 6-day life cycle, the PMNs migrate through the tissues and eradicate all external threats via degranulation.27 In the case of PMN activation by any trigger, including the AT1R pathway, NADPH oxidase initiates the oxidative burst by creating highly reactive superoxide radicals from molecular oxygen, which are then converted by superoxide dismutase into hydrogen peroxide, followed by the creation of hydroxyl ions, potent free radicals, via the Fenton reaction. Another by-product of the cascade is the hypochlorite radical, formed by the conversion of hydrogen peroxide into hypochlorite ion by myeloperoxidase (MPO).28 The free radicals destroy cell membranes and DNA, resulting in activation of poly(ADP) ribose and the exhaustion of intracellular ATP stores. In addition, the free radical burst results in an increase in intracellular ionised iron and calcium concentrations, causing the destruction of many biomolecules (Figure 1).20
Free radicals also affect cellular phospholipid metabolism, initially in the endothelium, the first point of contact of PMNs with tissues, resulting in the enhanced synthesis of lipid mediators, specifically leukotriene B4 (LTB4) and the platelet-activating factor (PAF), both of which are adept at promoting the further chemotaxis of phagocytes. In addition, intracellular xanthine oxidase generates additional superoxide radicals from xanthine, and the superoxide anions activate phospholipase A2, resulting in a notable change in arachidonic acid metabolism that leads to considerable changes in leukotriene and prostaglandin (PG) synthesis. The hypochlorite anion forms a protein–amino–hypochlorite after reacting with the free amino terminals of proteins. The protein–amino–hypochlorite inactivates nitric oxide (NO), a potent PMN inhibitor. In addition, the hypochlorite activates the proteases released from the azurophilic granules of PMNs.20,28 The decreases in NO lead to vasoconstriction and microvascular blood stasis, initiating microthrombosis.20
PMNs and MPHs share a common dependent pathway, and the IL-8 produced facilitates the evolution of immature neutrophils into functionally active granulocytes. MPHs cells behave in a similar manner to PMNs during their maturation, diapedesis and migration to the tissues; they also phagocytose foreign particles and release soluble factors.27,28 Autocrine, paracrine and endocrine mechanisms coordinate the regulation of the phagocyte system. The primary regulators are the cytokines, including IL-1, IL-6 and interferon-γ. The expression of histamine and catecholamine receptors on the phagocytes reveals a further neuroendocrine regulatory pathway of this defensive system. In contrast, glucocorticoids effectively suppress both the destructive biosynthetic processes and phagocytosis.20,28 Disproportionally high phagocyte activation in patients with severe COVID-19 results in ‘hit-and-run’-type damage primarily in the heart and lungs, making therapeutic outcomes less favourable.20
The typical sites of PMN/MPH extravasation are in the post-capillary HEV. The local vascular smooth muscle cells secrete NO, IL-1 and IL-6, thereby activating the HEV. In addition, the expression of PAF and P-selectin on the endothelial cellular surface intensifies within minutes of HEV activation.20 After the PMNs adhere to the endothelial cellular surface, the endothelium secretes IL-8 and expresses E-selectin after some hours to days; meanwhile, phosphorylation of lymphocyte function-associated antigen (LFA)-1 leads to G-actin polymerisation and the production of F-actin required for phagocyte diapedesis.20,29,30 Experimental polysaccharide blockade of selectins limited myocardial reperfusion injury, revealing the destructive nature of phagocyte overstimulation in the involved tissues.20,31
P-Selectin molecules are excessively expressed in diseased organs, but not in ‘resting’ endothelium, and play a role in the homing and migration of phagocytes.29 Furthermore, activated granulocytes may damage the endothelial cellular surface, triggering the coagulation cascade.32–35 In addition, the free radicals have a prothrombotic effect, promoting platelet aggregation by modifying prostacyclin synthase function and inhibiting antithrombin III.36,37 Thrombin is an effective P-selectin activator, so a vicious cycle of phagocyte attraction, endothelial damage, local coagulation and PAF-triggered coagulation on the HEV endothelial cellular surface may explain the predisposition to microthrombosis described in disease-affected organs in severe COVID-19 infections (Figure 2).38,39
Coronavirus Infection and Established Ischaemic Heart Disease
The effects of phagocytes and free radicals on ischaemic heart disease, especially reperfusion injury and arrhythmias, is well-established. However, the impact of SARS-CoV-2 infection on the heart is not yet entirely known, although there are reports that SARS-CoV-2 infection can lead to myocarditis and may exacerbate pre-existing ischaemic heart disease.40,41 As discussed in the previous section, virus-activated leucocytes can cause direct tissue damage or lead to end-organ microthrombosis, a critical phenomenon in the acute manifestation of chronic, stable ischaemic myocardial disease.38,39
Both experimental and clinical investigations have revealed significant effects of free radicals and phagocyte activation in unstable angina. P-Selectin expression is increased in atherectomy specimens from patients with unstable angina, predominantly in the endothelium. In addition, the expression of cellular adhesion molecules on circulating phagocytes and soluble P-selectin levels are markedly elevated in unstable angina, and increased expression of the adhesion molecule MAC-1 was detected on phagocytes in blood samples collected from the coronary sinus.30 In a canine model, histological samples of reperfused ischaemic heart muscle in the initial hour of reperfusion contained CD64-positive monocytes in the HEV and perivascular connective tissue.42 In addition, the phagocyte count in the extracellular fluid of the canine heart was elevated from 1 to 4 hours after successful reperfusion.42 Initially, the PMN proportion was dominant, but this shifted to a predominance of MPH by 4 hours.42 The initial chemotactic activity results from C5a then shifts to TGF-β1, which is coordinated by the monocyte chemoattractant protein (MCP-1).43 These pathways are crucial in repairing ischaemic damage; however, if they become dysregulated, they can lead to further myocardial injury, which could be the case in fulminant SARS-CoV-2 infections. The hypertensive excess resulting from viral ACE2 blockade may also trigger acute angina or acute heart failure (HF) in some individuals with established coronary disease (Figure 2).
Emerging data from the pandemic indicates that an over-reactive immune response may be seen in severe or fatal outcomes.23,24 The phagocytes first attack the microvasculature binding to the HEV and then cause destruction in the affected tissue, not only at a cellular level but also in the intercellular space. The initial 4- to 6-day ‘honeymoon period’ in mildly symptomatic SARS-CoV-2 infection and the sudden escalation in respiratory compromise parallels the tissue lifecycle of phagocytes. If the activation of these cells is limited because of a lower viral load, for example, or the host immune reaction/RAS response remains within the physiological range, a smooth recovery may be more likely. In progressive infections, microvascular thrombosis is the first event of the process (Figure 2).20 Procoagulant activity has been described in SARS-CoV-2-infected lung tissue samples containing diffuse microthrombi.44 Therefore, early anticoagulation is essential in patients with elevated coagulation markers; a relatively increased cumulative incidence of venous thromboembolism was reported in the absence of typical acute disseminated intravascular coagulopathy in COVID-19 pneumonia, even when patients were administered pharmacological thromboprophylaxis.45,46 Furthermore, heparin binds to the viral S-proteins and downregulates IL-6, supporting its early administration during the course of infection. Based on these data, numerous proactive approaches using early therapeutic anticoagulation or intensified pharmacological prophylaxis have been suggested.20,47 However, more data and prospective trials are needed to establish the role of anticoagulation in mitigating the coagulopathy associated with the COVID-19 pandemic.
There are reports that even minor plaques can result in adverse coronary events, mainly in combination with the prothrombotic status caused by the coronavirus disease.41 Aggressive antithrombotic therapy and/or prompt coronary revascularisation may provide a satisfactory solution; in case of failed therapeutic attempts, mechanical circulatory support is the bailout.48 It has been suggested that the timing of surgical interventions, including cardiac surgery, be postponed by 7 weeks after the initial SARS-CoV-2 infection due to the increased postoperative morbidity and mortality of patients with active COVID-19 disease at the time of surgery, although, of course, this decision must be balanced by the urgency of the condition requiring surgical intervention.49 Several risk factors associated with adverse outcomes with COVID-19 are potentially modifiable. Primary and secondary prevention strategies that target cardiovascular risk factors may improve outcomes for people with COVID-19.50 Therefore, maintaining follow-up for chronic heart disease is essential in protecting the population.
Despite the virus-derived leucocyte activation and risk of exacerbation of pre-existing ischaemic heart disease, the number of cardiac emergencies decreased in numerous countries during COVID-19 lockdown periods. As a consequence of curfews, many patients with chronic coronary disease stayed home and experienced lower stress levels than usual, or did not exercise at all, resulting in reduced provocative effects on their existing coronary burden. This observation is reinforced by a large-scale database analysis from France, in which a progressive reduction in ambulatory blood pressure values was registered compared with the pre-lockdown era.51 In addition, there may have been a fear component involved in the reduction in cardiac emergencies, whereby people did not seek medical attention for complaints relating to cardiac symptoms.52 However, patients experiencing ST-elevation MI who were admitted during the first wave of the COVID-19 pandemic experienced longer total ischaemic times, resulting in a more severe disease status upon hospital admission and a higher rate of in-hospital adverse events compared with a comparable pre-COVID-19 period.40,41,52 These adverse outcomes resulted from multiple confounding factors, including viral infection triggering additional ischaemia, healthcare systems that were overwhelmed by the pandemic and a delay in procedures following admission because of the need for precautionary infection control measures.
Myocarditis
One-quarter of patients hospitalised with COVID-19 experience additional myocardial involvement, with a smaller proportion being caused by direct cytopathic, myocarditic changes. The possibility of autoimmune reactions in this process has been raised.53 There are insufficient data as to whether successful acute treatment of the infection affects long-term myocardial recovery and cardiac pump function. Some patients experience subclinical cardiovascular abnormalities, and even those with recovered heart function may remain at risk of delayed cardiomyopathy and cardiac arrhythmias (Figure 2). The most appropriate long-term follow-up cardiac testing, also taking into account cost-effectiveness, for post-COVID-19 myocardial dysfunction and arrhythmias must be established. Standard ECG and echocardiogram 1 and 6 months after recovery, similar to cardiosurgical postoperative follow-up, may be reasonable. However, even these tests may not reveal subtle clinical abnormalities, and advanced imaging, including magnetic resonance imaging with gadolinium enhancement or an echocardiographic strain test, may be required in selected cases when initial testing reveals abnormalities, or as clinically indicated.54,55
Takotsubo syndrome is caused by a catecholamine surge, which may occur in COVID-19 disease due to the cytokine storm. Despite a lower prevalence of cardiac comorbidities in COVID-19-infected patients with takotsubo syndrome, direct myocardial injury, stress and inflammation may result in takotsubo syndrome with a high complication rate, although most patients recover successfully; in addition, this patient population is predominantly composed of elderly females.56
Cardiac Arrhythmias
Cardiovascular involvement in COVID-19 may result in arrhythmias. Offerhaus et al. analysed 5,782 hospitalised COVID-19 patients, finding that 11.0% experienced AF during hospitalisation and 1.6% experienced atrial flutter (AFL).57 Ventricular arrhythmias were observed in <0.8% of patients, and a conduction disorder was found in 6.3%. New-onset AF/AFL was associated with increased in-hospital mortality, mainly in males aged 60–72 years.57 In another analysis, the left atrial volume index and left ventricular mass index were significantly increased in patients with cardiac injury who had COVID-19 pneumonia, with an AF occurrence of 40.0%. These patients required invasive or non-invasive mechanical ventilation and had a higher mortality rate. Arrhythmias are multifactorial and complex in this patient population and result from hypoxia, acidosis, metabolic derangements and neurohormonal and catecholaminergic stress. Pathophysiological mechanisms of arrhythmogenesis include spontaneous release of calcium from the sarcoplasmic reticulum, autonomic nervous system-induced calcium entry into cardiac myocytes and possible direct atrial injury resulting from coronary artery disease accompanied by small vessel thrombosis.58
Ventricular arrhythmias affect a modest proportion of SARS-CoV-2-infected patients, although occasionally represent the only initial symptom. QT prolongation inconsistently predicts ventricular arrhythmias; in addition, hydroxychloroquine is seldom associated with malignant arrhythmias (<1%). ST-T wave changes are not clearly related to ventricular arrhythmias; nevertheless, elevated systemic inflammatory and cardiac injury markers are significantly higher in individuals presenting with life-threatening arrhythmias.59
There is a correlation between COVID-19 and new-onset atrioventricular (AV) blocks, but direct causality cannot be established due to various confounding factors. New-onset AV block is often benign and likely does not affect immediate and long-term clinical outcomes; the decision to undertake appropriate rhythm follow-up should be considered on a case-by-case basis.60 ECG features of SARS-CoV-2 are not characteristic, especially during the initial disease phase. ECG changes are usually late onset and do not occur in parallel with pulmonary abnormalities. Echocardiography may be a useful tool to distinguish between COVID-19-related myocardial damage and primary cardiac disease, and to monitor and assess COVID-19-related cardiovascular complications.61
Heart Failure
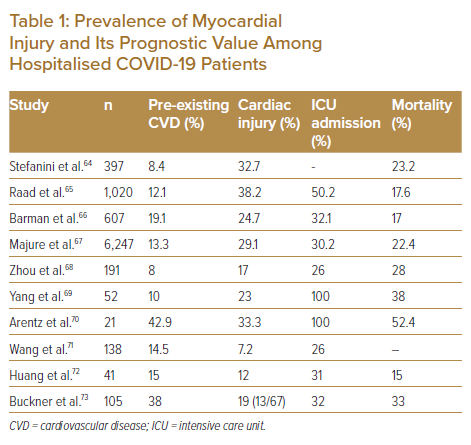
HF is a common condition that may develop at different stages during the course of a SARS-CoV-2 infection; severe inflammatory reaction exaggerates pre-existing myocardial disease.62 In combination with the higher metabolic demand due to sepsis, this can cause myocardial depression and either new-onset HF or acute decompensation of chronic HF. Another contributor to myocardial depression could be the coagulation dysfunction induced by sepsis.62 Elevated natriuretic peptides suggest HF with a worse prognosis in COVID-19 and warrant at least an echocardiogram to further assess cardiac function.63 Cardiac markers, such as troponin and creatine kinase-MB, increase significantly more in patients with cardiovascular risk factors and are associated with notably higher morbidity and mortality.40,50 Table 1 presents the prevalence of myocardial injury, and its prognostic value, in several studies in hospitalised COVID-19 patients.64–73
In the initial months following the emergence of the COVID-19 pandemic, several repurposed medications, such as hydroxychloroquine, antimicrobials and antivirals, were touted as potential treatments for the SARS-CoV-2 infection. This raised concerns regarding pharmacodynamic and pharmacokinetic drug-drug interactions, mainly involving concomitant QTc-prolonging agents and cytochrome P450 inhibitors.51,74 This is particularly relevant in patients with existing HF or after transplantation, given that these patients are likely to be on medication with the potential for significant interactions. As more data emerged that showed such treatments to be ineffective, the use of these repurposed agents and therefore the aforementioned concerns have subsided. However, QTc prolongation continues to be an essential aspect of drug monitoring for all hospitalised patients and should not be ignored despite the decline in the use of ‘red flag’ medications such as hydroxychloroquine, lopinavir/ritonavir and azithromycin in this particular setting. Evaluating the baseline risk of QTc prolongation for all hospitalised SARS-CoV-2 patients using the Tisdale risk score could offer insights into an individual patient’s risk of this complication and the subsequent risk of torsade de pointes.75
The emergence of the SARS-CoV-2 also led to early speculation regarding the chronic implications of certain cardiovascular medications, including ACE inhibitors, in relation to the infection. These medications are the backbone of guideline-directed medical therapy for HF. Patients with chronically upregulated ACE2 receptors, such as those on ACE inhibitors, have been shown not to have an increased risk of viral uptake. Cardiovascular societies issued statements early on during the pandemic to correct this perception of increased risk and avoid having patients stop these medications when they would otherwise benefit from evidence-based therapies. High-quality evidence later emerged from clinical trials, including REPLACE COVID and BRACE CORONA, providing assurance that these medications are safe and should not be suspended in patients hospitalised with mild or moderate COVID-19, which is consistent with the recommendations of international societies.76,77 Nonetheless, the practice of withholding RAS inhibitors in patients who develop systemic hyperinflammation and progress to vasoplegic shock should continue to be implemented in the setting of severe COVID-19, similar to other scenarios in critically ill patients.
If the developing cardiogenic shock necessitates advanced therapy, this means venoarterial (VA) extracorporeal membrane oxygenation (ECMO) rather than venovenous (VV) extracorporeal ECMO in COVID-19 respiratory distress. It was recently reported that in case of an escalation of therapy from VV- to VA-ECMO, a relatively low-flow VA perfusion was required, highlighting the efficacy of such mild circulatory support without inducing left ventricular distension in patients with COVID-19-related cardiopulmonary shock.78 The timing of care escalation is crucial to achieving improved outcomes. COVID-19 patients on mechanical ventilation with increased positive end-expiratory pressure (PEEP) may have compromised right ventricular filling, resulting in extra fluid resuscitation or pressor support. Therefore, lung-protective ventilation is recommended with an early escalation strategy. In the literature, high pre-ECMO PEEP (>15 cmH20) on ventilator and low respiratory system compliance (<30 ml/cmH20) are independent predictors of mortality, and delaying tracheostomy is an additional risk factor for death. The inflammatory burst and concomitant procoagulative status may significantly influence outcome, an elevated pre-ECMO C-reactive protein level is an additional risk factor for mortality and the required ECMO flow is higher in non-survivors than survivors.79 In severe SARS-CoV-2 respiratory failure, a long period of invasive ventilation and extracorporeal support is often required. There is a non-negligible burden of thromboembolic disease and renal injury associated with ECMO treatment. A large proportion of patients need tracheostomy and steroids to facilitate the challenging weaning process.80 Therefore, a thorough risk–benefit analysis of escalated therapy for each candidate is suggested so that patients with an increased probability of survival can benefit from scarce regional resources during the pandemic.81
One report compared the 6-month mortality of patients receiving VV-ECMO support for COVID-19 with a historical viral ARDS cohort.82 Patient characteristics, ECMO parameters, complications and mortality were compared between the two cohorts. At 6 months, survival was significantly higher in the COVID-19 than in non-COVID-19 viral pneumonia group. Patients with COVID-19 who survived to decannulation had a higher Murray score, decreased burden of organ dysfunction, higher incidence of pulmonary embolism and longer ECMO runs than those who did not. However, survival in patients on ECMO for COVID-19 was similar to those treated for non-COVID-19 viral ARDS.82
Cellular immunity is altered in long-term ventricular assist device (VAD) recipients.83 However, there is no evidence that these patients have a higher risk of acquiring SARS-CoV-2 infection. Although cardiac output is maintained at a constant level by the VAD, optimal preload and afterload are essential to keep actual output steady, even in the context of a systemic infection. If haemodynamics are compromised, various VAD-associated complications may occur, including low-flow and suction events, concomitant pump thrombosis or right ventricular failure. Studies have reported similar complications and risk profiles for COVID-19-infected VAD recipients and patients receiving ECMO.84
In heart transplant patients, emerging infections are always a concern given life-long immunosuppression to prevent rejection and preserve graft function. Although immunosuppression elevates susceptibility to viral infections such as herpes or cytomegalovirus, acute respiratory viral infections are uncommon. SARS-CoV-2-infected heart transplanted patients share similar demographic and clinical features with the general population regarding male predominance, comorbidities and clinical presentation. Whether heart transplanted patients are more prone to acquiring COVID-19 remains unclear. Furthermore, most of these patients presented with absolute lymphopenia, a common finding in SARS-CoV-2 infection, and this is independently associated with higher morbidity and mortality. However, myelotoxicity related to the immunosuppressive treatment in transplant recipients may also result in a similar picture. Modification of the baseline immunosuppressive regimen is diverse in the literature. The American Society of Transplantation suggests reducing immunosuppressive therapy with the maintenance of corticosteroids in SARS-CoV-2-infected patients who have not had recent rejection episodes, especially in severe COVID-19 cases.85,86 The International Society for Heart and Lung Transplantation (ISHLT) has similar recommendations for moderate to severe COVID-19.87 Regarding patients recovering from COVID-19 on the waiting list, it is essential to obtain two negative polymerase chain reaction tests 2 weeks after the initial SARS-CoV-2 diagnosis in order to proceed with transplantation depending on the acuity of the disease and organ availability. The ISHLT recommendations support testing for the SARS-CoV-2 in donors, if available, and donors should be rejected in case of positive testing and imaging signs of viral pneumonitis.87 Moreover, due to the possibility of nosocomial cross-infections, a second test for donors who previously tested negative should be considered before organ harvesting.62
A similar trend was observed among HIV-positive patients; the absolute cumulative COVID-19 mortality was low, at <0.1%, reflecting the young age profile of this population. Conversely, patients with comorbidities show an increased death rate compared with the general population. The association between HIV and COVID-19 mortality is more pronounced among people of African origin, with a 4.3-fold higher risk of COVID-19 death in this group. Nevertheless, this group is also at increased risk of adverse HIV outcomes, including virological rebound.88
Vaccination and the Heart
Vaccines against SARS-CoV-2 infection are effective within clinical trials of immunocompetent people, significantly reducing severe disease probability, hospitalisation and mortality. Most vaccines use viral mRNA to replicate the S-protein subunits of SARS-CoV-2 to hinder viral entry and stimulate both cellular and humoral immune responses. Nevertheless, some studies have demonstrated low or nearly no response to vaccination in heart transplant recipients, although immune paresis, a weaker cellular and humoral response than expected to an antigenic stimulus in heart transplant hosts is not unique to the COVID-19 vaccine in single-organ-transplanted recipients; hence, lower rates of immune response have been reported with other vaccines in this vulnerable cohort.89
Myocarditis is a rare complication of COVID-19 mRNA vaccinations, commonly occurring in young adult men. The incidence of myocarditis measures 12.6 cases per million second-dose mRNA vaccinations in those aged 12–39 years. Patients with post-vaccination myocarditis invariably present with chest pain accompanied by elevated cardiac troponin levels, usually 2–3 days after the second vaccination dose. ECG abnormalities with ST elevation are common; cardiac MRI is suggestive of myocarditis in all affected patients. In some cases, autoantibody titres against certain self-antigens and levels of natural killer cells are increased. The mechanisms underlying the development of myocarditis are unclear; triggered pre-existing dysregulated immune pathways in certain individuals, a hyperactive immune response to mRNA and dysregulated cytokine flair-up have been assumed. The reason for male predominance is also unknown; a possible explanation may rely on sex hormone-influenced differences in immune response, and an underdiagnosis of cardiac disease in women may also contribute. Almost all patients show resolution of signs and symptoms, as well as improvements in diagnostic markers and imaging studies, with or without treatment.90
Cerebral microthrombotic activity has been reported at a very low incidence in freshly vaccinated people, mainly on the venous side. The thrombotic tendency results predominantly from the phagocyte activation cascade and free radical discharge. In strong responders on the RAS–phagocyte activation pathway, the S-protein trigger itself may ignite the microthrombotic tendency. Therefore, additional low-dose post-vaccination acetylsalicylic acid therapy for 1 week could be beneficial in avoiding this complication, and the risk of this brief therapy is negligeable.20
Innate immunity triggers responsible for infection limitation or a hyperinflammatory response in SARS-CoV-2 infection are primarily uncharted territories. The SARS-CoV-2 S-protein primes inflammasome formation and the release of mature IL-1β from macrophages in COVID-19 patients, but not from macrophages in healthy SARS-CoV-2-naïve individuals. S-protein-driven inflammasome activation in macrophages from convalescent COVID-19 patients with distinct epigenetic and gene expression signatures is suggestive of innate immune memory after recovery from the SARS-CoV-2 infection. S-protein-driven IL-1β secretion from macrophages demands non-specific monocyte preactivation in vivo to trigger NOD-, LRR-, and pyrin domain-containing protein (NLRP3)-inflammasome signalling. The NLRP3 inflammasome is a multiprotein complex required for the secretion of the proinflammatory cytokine IL-1β that is crucial in COVID-19 hyperinflammatory syndromes. The SARS-CoV-2 S-protein, even as a vaccine antigen, triggers NLRP3 inflammasome activation and cytokine secretion in COVID-19 patient-derived macrophages. SARS-CoV-2 infection results in the reprogramming of human macrophages that enables rapid inflammasome assembly. Profound changes in macrophage gene activation and expression for several weeks to months after infection in COVID-19 patients may facilitate a better understanding of post-COVID-19 inflammatory syndromes.91
Supportive Therapy
Early introduction of an angiotensin receptor blocker to therapy, as long the patient’s blood pressure asymptomatically allows (≤110 mmHg), may result in favourable outcomes by weakening the cytokine burst.92–94 Microthrombotic activity on a capillary level requires low-molecular-weight heparin prophylaxis even in the early phase of the disease.95 Additional antiplatelet therapy must be considered because the thrombotic activity is caused by the phagocyte activation cascade and free radical burst.32,33
Remdesivir was developed for Ebola virus infections and is a monophosphoramidate nucleoside prodrug that can easily penetrate the cell membrane. Upon entering target cells, remdesivir monophosphate is rapidly converted into its active triphosphate, and, in RNA viruses, it acts as a substrate for the viral replicase, whereby it competes with endogenous ATP for incorporation in elongating RNA strands. After its incorporation, remdesivir triphosphate causes synthesis arrest by inducing delayed chain termination.96 Thus, remdesivir improves recovery from SAR-CoV-2 infection in hospitalised patients and reduces morbidity, mortality and time to clinical improvement. Furthermore, in patients who do not require mechanical ventilation or ECMO, a 5-day course of remdesivir may provide similar outcomes and fewer side effects than double-length treatment.97
Molnupiravir is another antiviral agent that has received emergency authorisation from the US Food and Drug Administration (FDA) by a marginal vote in its favour. The initial Phase III trial showed that molnupiravir reduced hospital admissions by 50%, but the data presented to the FDA revealed only a 30% admission rate reduction. Molnupiravir triggers an accumulation of errors in the viral genome during the replication phase, and this raised a concern regarding the possibility of human genome alteration. However, animal studies revealed a low risk for adults at therapeutic doses.98
Another antiviral agent, PF-07321332, a 3-chymotrypsin like protease (3CLpro) inhibitor targeting the main SARS-CoV-2 protease essential in viral replication, is currently in phase III trials in combination therapy with ritonavir. The scheduled interim analysis in the trial demonstrated an 89% reduction in the risk of COVID-19-related hospitalisation or death from any cause compared with placebo-treated study subjects within 3 days of symptom onset.99
A combination of the monoclonal antibodies imdevimab and casirivimab is also a promising treatment strategy for SARS-CoV-2. This combination drug has effectively reduced both the number of medical visits and the viral load in COVID-19 patients, and shows in vitro activity against current concerning variants of the virus. The main limitation of this therapy at the moment is primarily financial.100
A further target in the fight against COVID-19 is in microvascular phase, by limiting the tissue damage caused by activated leucocytes. However, this therapy is not microbe specific and effective in any vasculitis accelerated on the phagocyte pathway. Vioprolide A is a natural product isolated from the myxobacterium Cystobacter violaceus. It is a highly potent substance, showing promising effects on the vascular endothelium in inflammatory processes by reducing phagocyte–endothelial cell interactions and targeting the importin family of carrier proteins without exerting cytotoxic effects. The inhibition of importin-dependent NFκB p65 nuclear translocation is crucial in the action of vioprolide A, resulting in reduced NFκB promoter activity and inflammatory gene expression.101
Limitations
This paper is a scoping review, providing an overview of the existing evidence in respect of adverse cardiac effects and relevant pathomechanisms involved in SARS-CoV-2 infection, regardless of the methodological quality of the included articles. In addition, we did not conduct a risk of bias analysis on the articles included. With ever-growing evidence regarding the COVID-19 pandemic, it remains challenging to provide a comprehensive and final overview relevant to daily cardiovascular clinical practice.
Conclusion
The SARS-CoV-2 pandemic represents a critical challenge to society worldwide over an extended period. Emerging experience reveals the microvascular target and multiorgan involvement of the infection, in contrast with the initial assumption that the respiratory system was the only target. Excessive activation of the RAS leading to an uncontrolled surge of the phagocyte system is the main driver of adverse outcomes, including short- and long-term cardiovascular complications. Furthermore, it has been established that complete restitution of integrity does not always commence after going through a SARS-CoV-2 infection, and that post-COVID-19 syndromes emerge in several organs, resulting in new or aggravating existing disease burdens.
Clinical Perspective
- Uncontrolled overstimulation of the phagocyte system results in microvascular disease in SARS-CoV-2 infection that may lead to unfavourable clinical consequences, including acute and chronic cardiac syndromes.
- Patients with STEMI hospitalised during the COVID-19 pandemic experienced longer total ischaemic time, resulting in a more severe disease status upon hospital admission and a higher rate of in-hospital adverse events compared with a parallel period. These adverse outcomes result from various confounding factors, including additional ischaemia triggered by the viral infection, overwhelmed healthcare systems during the pandemic and a delay in procedures because of precautionary infection control measurements on admission.
- HF may develop at different stages during the course of a SARS-CoV-2 infection; the severe inflammatory reaction can exaggerate pre-existing myocardial disease. Therefore, regular follow-up on established HF patients, even in the form of remote videographic consultations, is essential during the pandemic.
- If developing cardiogenic shock necessitates advanced therapy, this means VA-ECMO in contrast to VV-ECMO in COVID-19 respiratory distress. In case of treatment escalation from VV- to VA-ECMO, a relatively low-flow VA perfusion is required, highlighting the efficacy of such mild circulatory support without inducing left ventricular distension in patients with COVID-19-related cardiopulmonary shock. Therefore, the timing of care escalation is crucial to achieving improved outcomes.
- Myocarditis is a rare complication of COVID-19 mRNA vaccinations, commonly occurring among young adult men, with an incidence of 12.6 cases per million second-dose mRNA vaccinations in those aged 12–39 years. Therefore, it is crucial to thoroughly investigate post-vaccination atypical chest discomfort in this at-risk population.