









The number of patients with heart failure (HF) is increasing worldwide due to ageing, and HF is one of the most common causes of death.1 Although there have been many studies on the mechanisms of HF, including abnormal calcium handling, adrenergic receptor overactivation, apoptosis and DNA damage in cardiomyocytes, the complex pathophysiology remains incompletely understood.2–6 The pathogenesis of HF has been extensively studied by many researchers with a focus on cardiomyocytes, but cardiomyocytes constitute only 20–30% of cells forming the heart, with >70% being non-cardiomyocytes, such as endothelial cells, fibroblasts, smooth muscle cells, pericytes and blood cells.7 This highlights the potentially pivotal role of non-cardiomyocytes in HF development.8
A failing heart is not an isolated entity. Haemodynamically, hypoperfusion and congestion induce dysfunction of extracardiac organs, such as the lung, liver and kidney. Elevated liver transaminase levels are associated with an increased risk of mortality, and the presence of chronic kidney disease is a potent poor prognostic factor for patients with HF.9,10 Renal dysfunction is well-known to be intimately related to HF, and albuminuria alone could provoke incident HF.11 Albuminuria is not only reflective of endothelial damage, but also precedes neurohormonal activation, including the renin–angiotensin–aldosterone system, and autonomous nervous system, which disturbs cardiac homeostasis. These results suggest that interorgan communications are deeply involved in the pathological conditions of HF.
The Roles of Non-cardiomyocytes in the Pathogenesis of Heart Failure
Endothelial Cells
The role of endothelial cells in cardiac function has been widely recognised.12 Endothelial cells form a single layer covering the blood vessels’ inner lining and perform many functions, such as the regulation of vasodilation, thromboprophylaxis, anti-inflammatory effects, antioxidant effects and neovascularisation. These functions are impaired in many diseases, including hypertension, obesity, diabetes and hyperlipidaemia, and in ageing individuals and smokers.13 Dysfunction of endothelial cells due to injury is known to be partially reversible, necessitating early therapeutic intervention through pharmacotherapy and lifestyle modification.
Heart Failure and Dysfunction of Endothelial Cells
Since the late 1980s, the concept that endothelial dysfunction in the heart is one of the causes of HF has gradually gained acceptance. Endothelial dysfunction refers primarily to abnormalities in the regulation of vasodilation and contraction, vascular permeability regulation, and inflammatory processes. These endothelial dysfunctions are assessed through peripheral blood flow-dependent vasodilation during congestion and through coronary flow reserve. In patients with cardiovascular diseases or those at risk, abnormal coronary flow reserve is strongly associated with increased risks of death and major adverse cardiac events.14
Endothelial dysfunction is believed to be deeply involved in the pathophysiology of cardiovascular diseases (CVD).15 One of the most crucial molecules responsible for endothelial function is nitric oxide (NO).16 NO is biosynthesised from arginine by the enzyme NO synthase (NOS). There are three isoforms of NOS: endothelial NOS, inducible NOS and neuronal NOS. Each is predominantly expressed in different tissues: endothelial NOS in vascular endothelial cells; inducible NOS in inflammatory cells, such as monocytes, macrophages and neutrophils, and neuronal NOS in the central nervous system. Normal endothelial cells release several relaxants, notably NO, which play a vital role in vascular regulation through guanylyl cyclase-cyclic guanosine monophosphate. Vasodilation is maintained by the continuous synthesis and release of NO, and vasodilation ability is reduced in patients with HF.17
Endothelial dysfunction is caused by an imbalance between endothelium-derived vasodilatory and vasoconstrictive actions, leading to reduced flow-mediated vasodilation and an inability to meet the oxygen demands of the heart. Additionally, endothelial dysfunction impairs antithrombotic activity and induces inflammation. There have been reports showing cardiac dysfunction in endothelial NOS knockout mice.18 In 2020, a phase III trial of the guanylyl cyclase stimulator, vericiguat, was conducted, showing a lower incidence of cardiovascular death and hospitalisation due to HF in high-risk patients treated with vericiguat.19 While the effects of vericiguat on various cells in the heart are known to be multifaceted, at least the improvement in vascular regulatory ability in endothelial cells is thought to contribute to these results.
Heart Failure and Senescence of Endothelial Cells
The relationship between vascular endothelial cells and ageing has been the subject of many studies.20 It has been reported that expression levels of p53 increase in the murine heart after pressure overload, and that p53 plays a critical role in the development of HF.5,21 In mouse models of diabetes, inhibition of p53 has been reported to prevent early-stage myocardial apoptosis and suppress diabetes-induced cellular senescence.22 Additionally, endothelial cell-specific knockout of p53 has been reported to improve cardiac function in a mouse model of pressure-overload HF, with one mechanism being the prevention of apoptosis.23 Another study has shown that heterozygous knockout of p21 in endothelial cells increases angiogenesis without increasing apoptotic cells.24 In mouse models subjected to ischaemia-reperfusion injury, the administration of the senolytic drug, navitoclax, reduced the area of MI.25 Furthermore, we have recently observed that administering CDK4/6 inhibitors to a mouse model of pressure-overload HF improves cardiac function by inhibiting excessive cell proliferation and senescence of endothelial cells. In conclusion, the senescence of endothelial cells is deeply linked to HF, and preventing vascular senescence may become a new therapeutic intervention for HF.
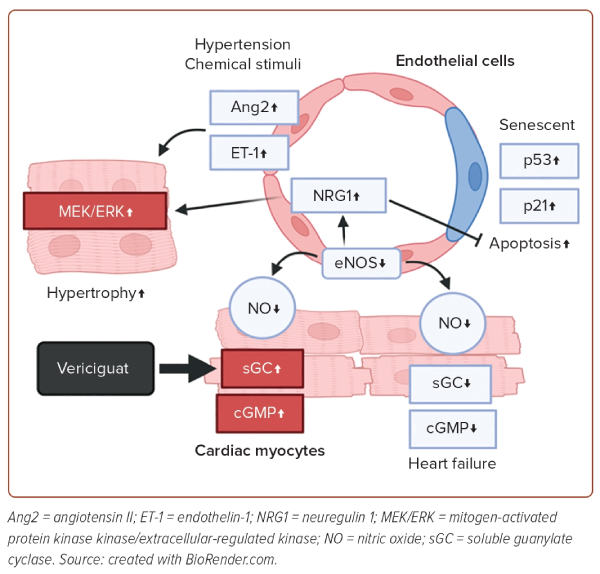
Heart Failure and Intercellular Communication
Substances that mediate intercellular communication are crucial for the crosstalk between endothelial cells and cardiac myocytes (Figure 1). Apart from NO, humoral factors, including angiotensin II, endothelin-1 and neuregulin-1, play pivotal roles in the crosstalk.26 Under various stimuli, such as mechanical stresses and chemical stimuli, endothelin-1 is secreted from endothelial cells, and induces hypertrophy of cardiac myocytes by activating the protein kinase C and mitogen-activated protein kinase kinase/extracellular-regulated kinase pathways, leading to the development of HF.27 Neuregulin-1, which is upregulated when NO levels decrease, induces cardiomyocyte hypertrophy, but protects cardiomyocytes by activating the erythroblastic leukaemia viral oncogene homologue pathway.28 These factors secreted from endothelial cells directly affect cardiomyocytes and are critically involved in the development of HF.
Thus, endothelial cells play pivotal roles in the pathogenesis of HF, not only by forming blood vessels that carry oxygen and nutrients, but also by secreting various factors. This suggests that targeting endothelial cells may offer a potential therapeutic avenue for HF. Various drugs are delivered to organs through the blood vessels, and since endothelial cells are located in the closest proximity to the blood, they are an organ that is easy to target for drug delivery. Given the ongoing poor prognosis associated with HF, the role of endothelial cells is increasingly viewed as a key target for the innovation of new treatments for HF.
Cardiac Fibroblasts
Cardiac fibroblasts constitute 10–15% of cardiac cells. In general, cardiac fibroblasts play a crucial role in fibrosis during wound healing in various organs and tissues by producing various extracellular proteins including collagens, which form the extracellular matrix.29 Cardiac fibroblasts also meticulously generate and arrange collagen to maintain extracellular matrix structure, creating a robust foundation in various situations, such as MI and mechanical stress. They play a vital role both structurally and mechanically in maintaining cardiac function. Due to the scarcity of reliable cardiac fibroblast markers, research on the role of cardiac fibroblasts was primarily confined to their mechanical functions. While markers, such as double data rate 2, ferroptosis suppressor protein 1, Thy-1 (CD90) and vimentin, were employed for fibrotic characterisation, their expression was also found in fibroblasts of other tissues besides the heart.
MI and Cardiac Fibroblasts
The identification of specific markers, such as Tcf21 and Pdgfrα, has propelled advancements in cardiac fibroblast research.30 The primary source of cardiac fibroblasts was identified as the epicardium during embryonic heart development. In hearts affected by MI and hypertrophy, however, the origin of activated fibroblasts was traced back to resident fibroblasts in the heart.31 Furthermore, the identification of periostin as the activated fibroblast marker has facilitated experiments with genetically engineered mice. These studies revealed transformations in cardiac fibroblasts, where resident fibroblasts differentiate into activated fibroblasts in the context of MI or HF. Subsequently, these activated fibroblasts further differentiate into myofibroblasts, contributing to a robust induction of fibrosis.32
Ultimately, in the setting of MI, cardiac fibroblasts undergo differentiation into matrifibrocytes, which lose their proliferative capacity and generate specific extracellular matrix constituent proteins, contributing to the stabilisation of the scar tissue (Figure 2). These findings suggest that cardiac fibroblasts alter their gene expression and morphology in MI to safeguard the heart.33 As mentioned earlier, research on cardiac fibroblasts has predominantly focused on the MI mouse model due to its notable phenotypic changes, and the role of cardiac fibroblasts in other heart diseases remains unclear. The relationship between cardiac dysfunction or the pathophysiology of HF and cardiac fibroblasts is not fully understood.
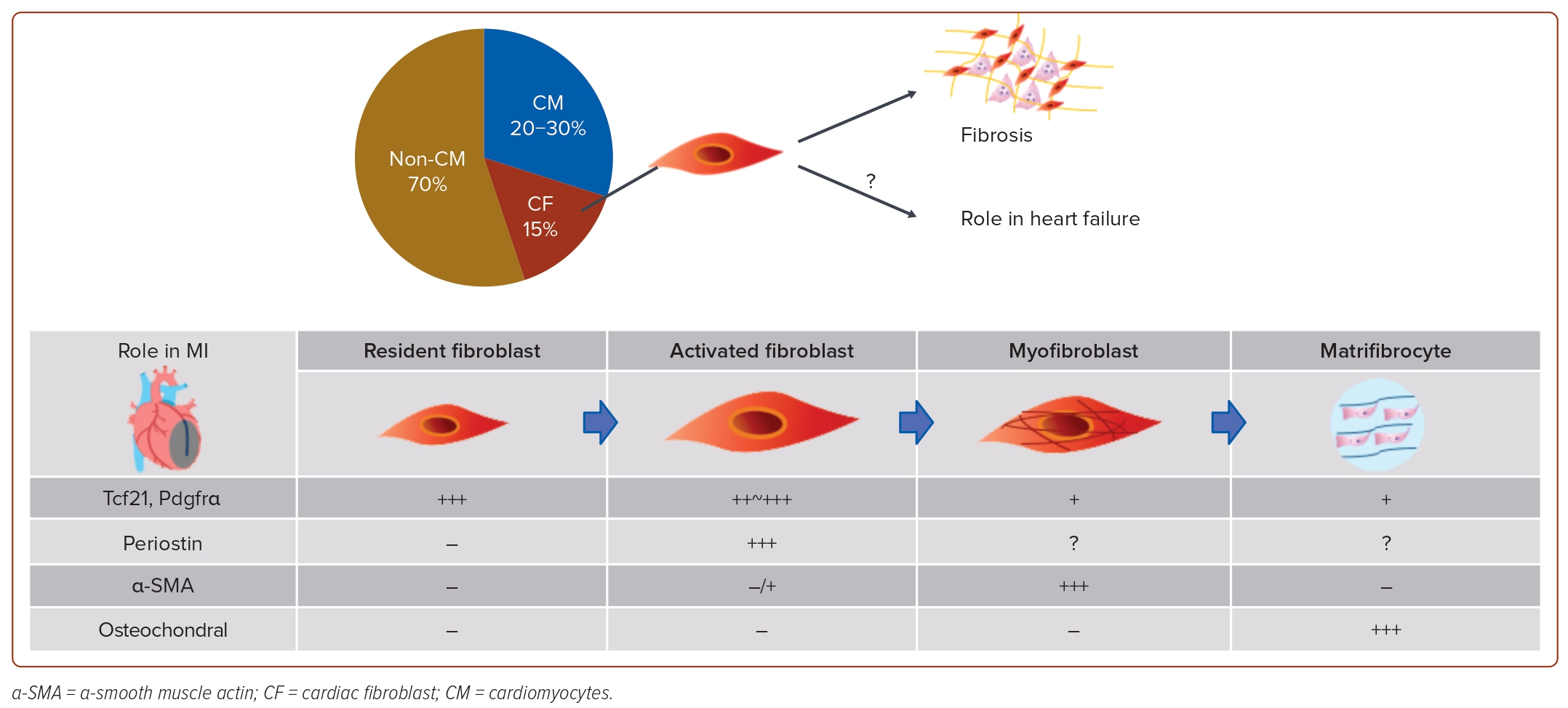
Heart Failure and Cardiac Fibroblasts
In the mouse model of HF induced by pressure overload through transverse aortic constriction, myofibroblasts were absent in the perivascular region, and only 15% of the total cardiac fibroblasts were found in the interstitial region 4 weeks after transverse aortic constriction.33 This result suggests that the function of cardiac fibroblasts in failing hearts differs from their role in stabilising scars by producing collagens in hearts with MI. In failing hearts, fibrosis is recognised to advance from the epicardium to the endocardium, driven by pressure overload-induced wall stress on the heart, microcirculatory disturbances and other factors.33
There are two patterns of fibrosis: one involves the accumulation of extracellular matrix around blood vessels, and the other entails the construction of fibrotic tissue among cardiomyocytes. While research on cardiac fibrosis has traditionally concentrated on mechanical changes, some recent studies have reported its involvement in the pathophysiology of HF. Although it is well-known that cardiac fibrosis is associated with left ventricular diastolic dysfunction, González et al. reported that an excess of fibrosis induces the remodelling of collagen fibres and cardiomyocytes, resulting in left ventricular systolic dysfunction.34 The same study also suggested that cardiac fibrosis could be a causative factor for arrhythmias, and that the accumulation of cardiac fibrosis might impede the delivery of oxygen to cardiomyocytes, leading to ischaemia.34 Another study indicated that eliminating cardiac fibrosis using chimeric antigen receptor T-cells resulted in improved cardiac function.35
While cell populations were once thought to be homogeneous and analysed collectively, the ability to analyse at the single-cell level has revealed significant heterogeneity within cell populations, even among cells of the same type.36 Using single-cell analysis, Ko et al. reported that high-temperature requirement factor A3 in cardiac fibroblasts regulates the amount of transforming growth factor-β (TGF-β), and that deletion of high-temperature requirement factor A3 induces not only marked fibrosis, but also cardiac dysfunction by activating TGF-β signalling and inducing DNA damage in cardiac myocytes.37 This study indicated that cardiac fibroblasts affect cardiac myocytes and play critical roles in the pathogenesis of HF, as well as fibrosis, and suggests that the interaction between cardiac fibroblasts and myocytes is a novel target for HF.
Blood Cells
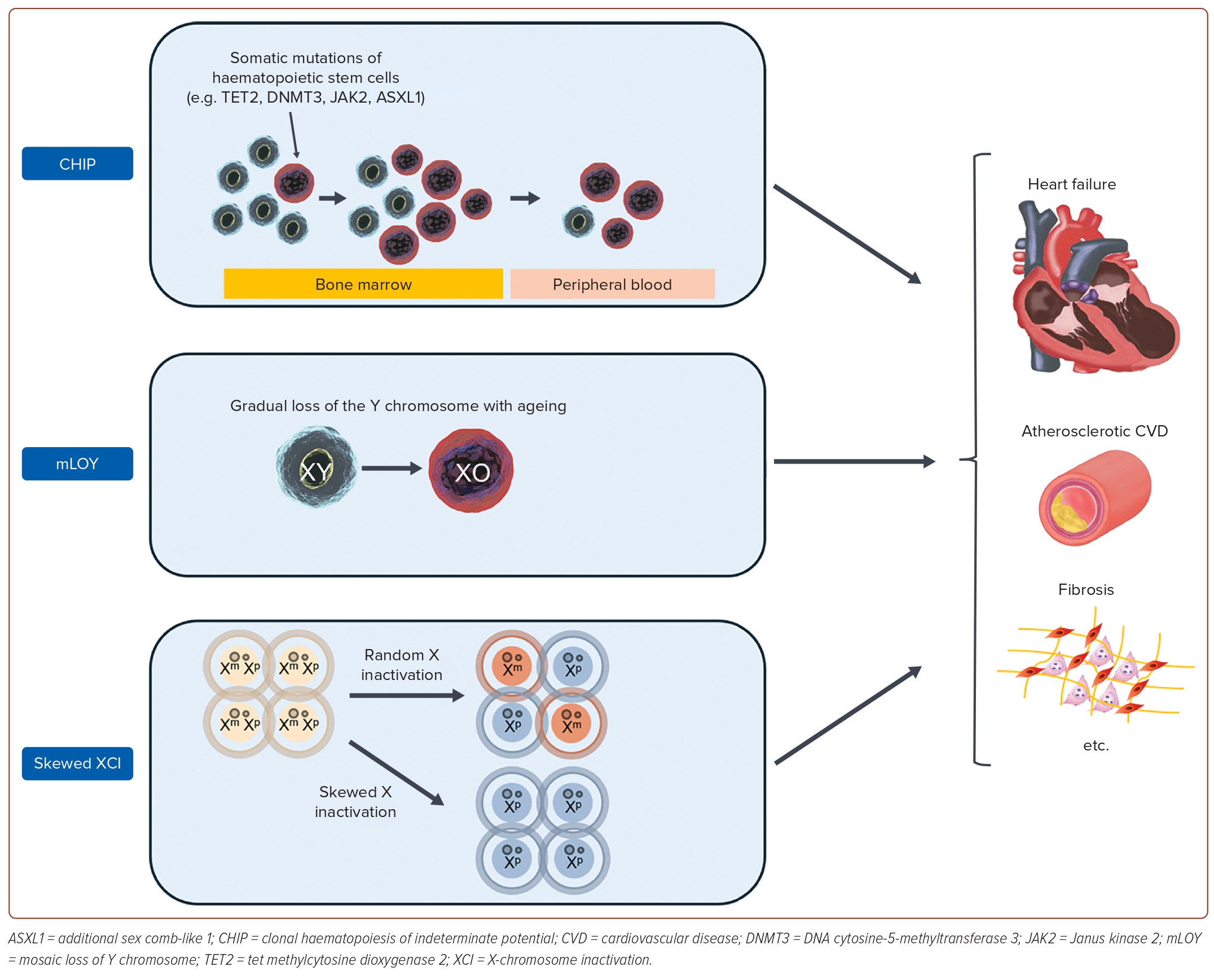
Growing evidence has shown a relationship between CVD and the immune system.38 The advent of next-generation sequencing has enabled the analysis of acquired mutations in blood cells that were previously not assessable by other techniques, such as flow cytometry or fluorescence microscopy. This has drawn attention to the association between somatic mutation cells and CVD, including HF. Here, we focus on the role of somatic mutations in haematopoietic stem cells in the onset and exacerbation of HF (Figure 3).
Clonal Haematopoiesis of Indeterminate Potential
Clonal haematopoiesis of indeterminate potential (CHIP), a premalignant state with somatic mutations of haematopoietic stem cells in the genes related to haematological malignancies, is associated with mortality due to various chronic diseases.39 Among them, CVD contributes significantly to the increased mortality from CHIP. The attention to the fact that CHIP worsens the prognosis of CVD began with a large study reported in 2014, which performed a whole exon analysis of approximately 17,000 patients.40,41 In this study, approximately 10% of patients aged >70 years had CHIP, indicating that CHIP is an independent risk factor for ischaemic heart disease and ischaemic stroke. Since then, many studies have examined the association between CHIP and CVD, and many basic studies have shown that CHIP induces inflammatory activation. For example, TET2 mutations induce secretion of interleukin (IL)-1β, a major proinflammatory cytokine, through activation of the nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 inflammasome and caspase-1, while DNMT3A and JAK2 mutations also induce excessive secretion of inflammatory cytokines, such as IL-6.42
In approximately 57,000 patients without established HF drawn from five European and US databases, a 25% increased risk of new onset of HF was reported in patients with CHIP compared with those without it.43 The study also showed that this risk was even higher in cases with larger clone sizes, and that the contribution of CHIP was dose dependent. In addition, this study also examined the differential contribution depending on which gene the mutation was in, and found that ASXL1 mutation-positive patients had significantly more left ventricular systolic dysfunction. In contrast, no such trend was observed for DNMT3A or TET2, suggesting that the significance may vary depending on the driver gene. More recent preliminary data have shown that different bases at the same locus of the DNMT3A mutation contribute differently to the development of CVD.44 Future studies that address the differences in effect size by gene and mutation are desirable.
Mosaic Loss of Y Chromosome
In males possessing one X chromosome and one Y chromosome, the gradual loss of the Y chromosome with ageing is referred to as mosaic loss of Y (mLOY). This phenomenon is primarily observed in blood cells, and its association with various chronic diseases has been reported in the literature.45,46 In cardiovascular diseases, it has been reported as an adverse prognostic factor in some conditions, such as cerebral infarction and post-transcatheter aortic valve replacement.47,48 Although studies on the association and mechanisms of mLOY with the development of HF are limited, Sano et al. suggested the following possibilities.49 The mice model with mLOY showed myocardial fibrosis, and the myocardial macrophages, induced by the loss of Y, were found to be in an activated inflammatory state. Additionally, inhibiting TGF-β1 was shown to alleviate myocardial damage, indicating the potential role of mLOY to induce HF through inflammation. Furthermore, using data from the UK Biobank, they demonstrated that men with mLOY had approximately a 30% increased mortality rate due to cardiovascular diseases over a tracking period of about 11 years. This suggests that mLOY could be a useful indicator for clinical stratification. Future studies are anticipated to investigate the role and effect size of mLOY in various CVDs.
Skewed X Chromosome Inactivation
X-chromosome inactivation (XCI) refers to the phenomenon in which one of the X chromosomes is silenced in each cell to regulate the expression levels of genes on the X chromosome.50 This is a phenomenon specific to females who possess two X chromosomes. Originally, XCI was believed to exhibit a 1:1 distribution in a cell population occurring randomly to maintain physiological homeostasis. However, it has become apparent that there is variation in some individuals.51 This phenomenon is referred to as skewed XCI. Roberts et al. analysed skewed XCI using whole blood samples from the TwinsUK registry, focusing on 1,575 women.52 This study quantified XCI, and demonstrated that XCI is an independent predictor of atherosclerotic CVD risk. Atherosclerotic CVD stands as a leading cause of HF, and activated inflammation has been reported to contribute to its pathogenesis. XCI has also been shown to induce an increase in monocytes and a decrease in interleukin-10 with anti-inflammatory properties, suggesting potential involvement in the progression of atherosclerotic CVD through inflammation. Additionally, Roberts et al. indicated that XCI serves as a risk factor for the onset of malignant tumours, implying potential involvement in major societal issues, such as HF and cancer.
The analysis of somatic mutations is expected to contribute to the realisation of further precision in medical care. Despite numerous studies conducted in recent years, it is important to note that these findings may undergo quantitative changes over time. Therefore, future research is eagerly awaited, particularly with the execution of longitudinal studies.
The Roles of Other Organs in the Pathogenesis of Heart Failure
Intercommunication Between the Heart and Kidney
Beneficial potentials of angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers and mineralocorticoid receptor antagonists for both HF with reduced ejection fraction and chronic kidney disease have been widely accepted. In addition, mounting evidence that sodium-glucose cotransporter 2 inhibitor (SGLT2i) improves the prognosis of both diseases has been building consensus.53 These findings indicate the existence of common pathophysiological mechanisms for HF and kidney disease. SGLT2i sustains favourable effects on failing hearts beyond short-term osmotic diuretic action. SGLT2i-induced ketogenesis promotes nutrient deprivation signalling, including activation of adenosine monophosphate-activated protein kinase and repression of mammalian target of rapamycin, which activates sirtuins and increases the expression of peroxisome proliferator-activated receptor γ coactivator-1α.54 Consequent enhancement of autophagic flux suppresses the development of cardiomyopathies.
SGLT2i also exerts direct cellular effects on cardiomyocytes and renal parenchymal cells regardless of the expression of SGLT2 by unknown mechanisms. Several papers reported the sympatholytic effects of SGLT2i presented as a repressed expression of tyrosine hydroxylase in kidneys.55,56
Regarding the renal sympathetic nerve traffic, the increased efferent signal in the setting of HF leads to inappropriate renal sympathetic tone, and upregulates renin–angiotensin–aldosterone activity and neprilysin expression in kidneys to cause excessive fluid retention, while the afferent sympathetic signal deteriorates cardiometabolic diseases in various ways.57 Augmented input signals from mechanoreceptors and chemoreceptors of injured kidneys are transmitted to the subfornical organ of the brain, which contains angiotensin II receptor type 1a-positive neurons regulating thirst and salt-appetite sensations.58 Subfornical organ neurons partly have connections with the paraventricular nucleus neurons projecting to the rostral ventrolateral medulla, a sympathetic nerve center.59 Kidney injury upregulates angiotensin II receptor type 1a signalling in the subfornical organ and subsequently activates the paraventricular nucleus and the rostral ventrolateral medulla.
Centrally integrated signals through the communications in the brainstem result in an increased sympathetic tone directed towards several organs and detrimental effects on failing hearts.60 However, sympathetic stimulation in kidneys also exerts an adaptive neuroimmune response for failing hearts. Efferent nerves stimulate renal collecting duct epithelial cells, which leads to the secretion of the cytokine, colony-stimulating factor 2.60 Circulating colony-stimulating factor 2 is essential for the maintenance of cardiac tissue macrophages. These tight cooperative communications among the heart, brain and kidney complicate the pathophysiology of a failing heart, and it could be attributed to inconclusive early studies of catheter-based renal denervation, although recent meta-analysis exhibited a beneficial potential of renal denervation for HF (Figure 4).61,62
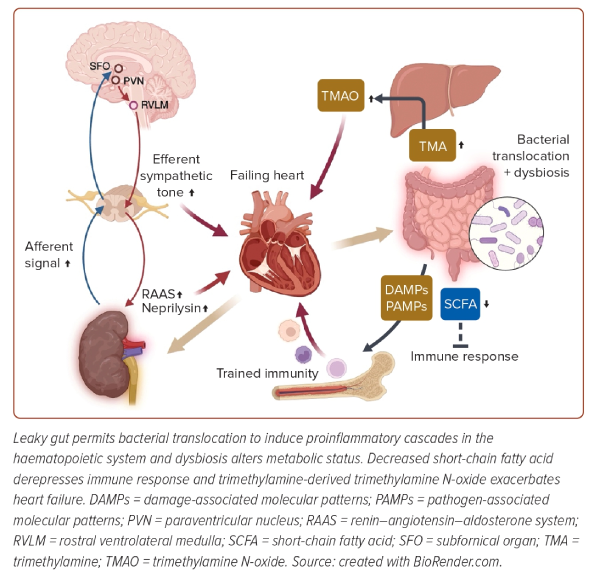
Intercommunication Between the Heart and Gut
Bowel wall oedema due to HF impairs intestinal barrier function, and a leaky gut allows bacterial translocation and the entry of damage-associated molecular patterns and pathogen-associated molecular patterns. The correlations of circulating endotoxin levels and intestinal epithelial cell injury markers with the severity of HF verify the gut hypothesis of HF.63,64 Failing hearts affect the diversity of gut microbial composition. Dysbiosis, a status of decreased intra-individual taxonomic diversity in HF, results in a decreased abundance of Roseburia, Faecalibacterium and Eubacterium rectale, known as butyrate-producing commensal bacteria.65 Short-chain fatty acids, including butyrate and acetate, do not only upregulate the expression of epithelial barrier genes, but also promote immune tolerance; thus, the alteration of microbiota in HF exacerbates inflammatory responses of leaky gut.
Regarding the metabolites produced by microbiota, trimethylamine N-oxide (TMAO) is also noteworthy. Dietary sources rich in choline, phosphatidylcholine and L-carnitine can be metabolised by gut microbiota to trimethylamine (TMA). TMA enters the portal circulation and is oxidised by hepatic flavin-containing monooxygenase into TMAO. While plasma TMAO activates platelets and causes endothelial dysfunction to facilitate atherogenesis, TMAO can directly induce cardiac dysfunction. TMAO activates TGF-β1 signalling to progress cardiac fibrosis.66 TMAO exacerbates mitochondrial function in failing cardiomyocytes via inhibition of the activity of pyruvate dehydrogenase.67 Mounting evidence supports the notion that elevated TMAO levels predict the poor prognosis of patients with HF, although the underlying mechanism has not been fully elucidated, and the clinical efficacy of reduction intervention remains elusive.67
Bacterial translocation and endotoxaemia unsurprisingly heighten inflammatory responses of the host immune system, and chronic low-grade inflammation contributes to the pathogenesis and progression of HF.68 Damage-associated molecular patterns and pathogen-associated molecular patterns stimulate pattern recognition receptors of innate immune cells and even cardiomyocytes to produce various inflammatory cytokines, and activate inflammasome to induce the secretion of IL-1β. IL-1β inhibits L-type calcium channels and desensitises β adrenergic signalling in cardiomyocytes, thereby weakening myocardial contractility, whereas IL-1 signalling decreases calcium reuptake into the sarcoplasmic reticulum to impair diastolic function.69 These mechanistic hypotheses are supported by the evidence that canakinumab, an IL-1β inhibitor, reduced hospitalisation and mortality of decompensated HF patients.70
Meanwhile, recent reports indicate that enhanced immune responses with accompanying epigenetic modulation and metabolic alteration in haematopoietic cells would be a central mechanism driving long-lasting inflammation.71 This trained immunity, or termed innate immune memory depending on the context, is implicated in the development of various CVDs, and especially centrally trained immunity at the level of haematopoietic stem cells (HSCs) in the bone marrow niche would be noticeable. Trained immunity in the setting of aging, referred to as inflammaging, is also triggered by translocations of gut microbiome-derived compounds, which stimulate myeloid cells in the bone marrow to sustain increased IL-1 secretion.72 IL-1 signalling drives preferential expansion of myeloid-skewing HSCs and imprints myeloid differentiation bias of HSCs. Periodontitis, which is also a risk of HF, permits microbial challenge in the circulation, and also upregulates IL-1β-mediated signalling in HSCs.73 Maladaptively trained HSCs could induce myelopoiesis and increased susceptibility to inflammatory comorbid conditions. Myelopoiesis and sustained inflammation could trigger incident HF, as exemplified by the association of CHIP and mLOY with a greater risk of HF, as discussed before.
Although the pathogenesis of HF has been extensively studied with a focus on cardiomyocytes, >70% of the cells in the heart are non-cardiomyocytes, and the interactions between many cell types, including cardiomyocytes and noncardiomyocytes, have been elucidated. HF is also a culminating status of miscellaneous heart diseases, and failing heart is closely intertwined with the pathophysiology of extracardiac organs. Therefore, this review was put together from a slightly different perspective than the more common traditional reviews, namely, the linkage between non-cardiomyocytes and cardiomyocytes, and between organs other than the heart and the heart. The approaches from these novel aspects may open new avenues for developing new treatments for HF. 
Clinical Perspective
- Heart failure is one of the most common causes of death.
- Non-cardiomyocytes play a pivotal role in the development of heart failure.
- Interorgan communication between the heart and other organs is important for heart failure.